|
|
|
Qu'est-ce que le longifolène ?
Le longifolène est un composé éthylénique appartenant à la famille des hydrocarbures terpéniques. Les terpènes sont des hydrocarbures polyinsaturés présents à l'état naturel dans les huiles essentielles extraites de certains végétaux.
On peut les considérer, d'un point de vue formel comme étant constitués par la répétition d'unités isopréniques : (C5H8)m
Le chimiste Ruzicka a proposé une nomenclature pour les terpènes en fonction du nombre d'atomes de carbone qui les constituent. On distingue les catégories suivantes selon la valeur de m :
|
m |
2 |
3 |
4 |
5 |
|
Nom |
Monoterpènes |
Sesquiterpènes |
Diterpènes |
Triterpènes |
|
|
|
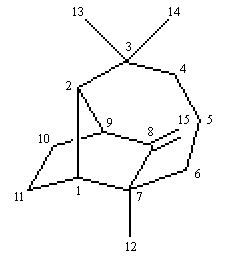
Molécule de longifolène
|
|
|
|
La molécule de (+)-longifolène, représentée à gauche comporte une structure tricyclique. Il s'agit du (1R,2S,7S,9S)-3,3,7-triméthyl -8-méthylènetricyclo-[5.4.0.02,9]undécane. Display : |
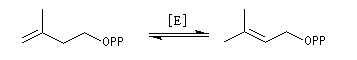
Structure du longifolène Biosynthèse du longifolène Le pyrophosphate d'isopentényle subit une isomérisation sous l'action d'une enzyme. Synthèses du longifolène racémique Stratégies possibles Les disconnections a et e conduisent à des structures de complexité moindre que la cible. En revanche, les disconnections b, c, d conduisent à des précurseurs à 8 chaînons difficiles d'accès.
La disconnection optimale reconnue par Corey correspond à la coupure de la liaison C2-C9 (voie a). C'est effectivement celle qu'il a utilisé dans sa synthèse historique [2], [3]. D'autres solutions plus audacieuses existent. Elles sont basées sur une double disconnection.
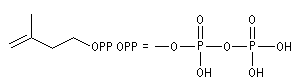
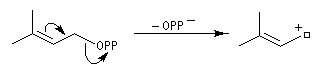
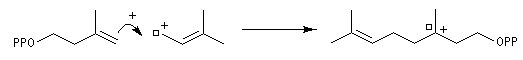
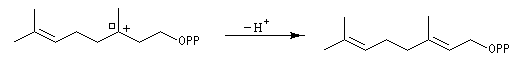
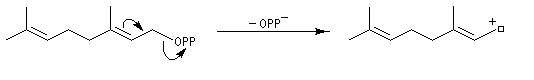
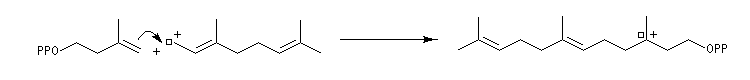
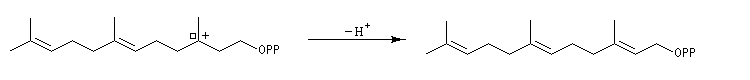
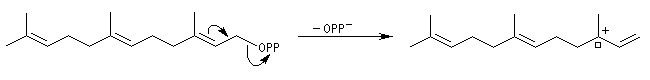
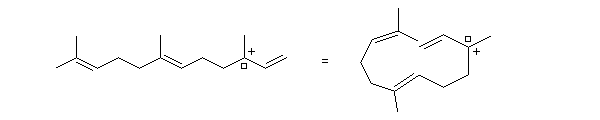
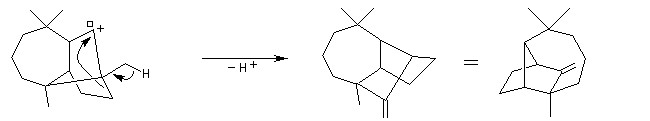
La première partie de la conférence prononcée par E. J. Corey à l'occasion de l'attribution de son prix Nobel de chimie en 1990 est consacrée à la synthèse du Longifolène. En effet, c'est Corey qui a le premier attiré l'attention sur le profit qu'on peut tirer d'une analyse disconnective d'une molécule cible afin d'établir une stratégie de synthèse optimale.
Si l'on examine les disconnections possibles à partir de la molécule de longifolène, on obtient le schéma suivant.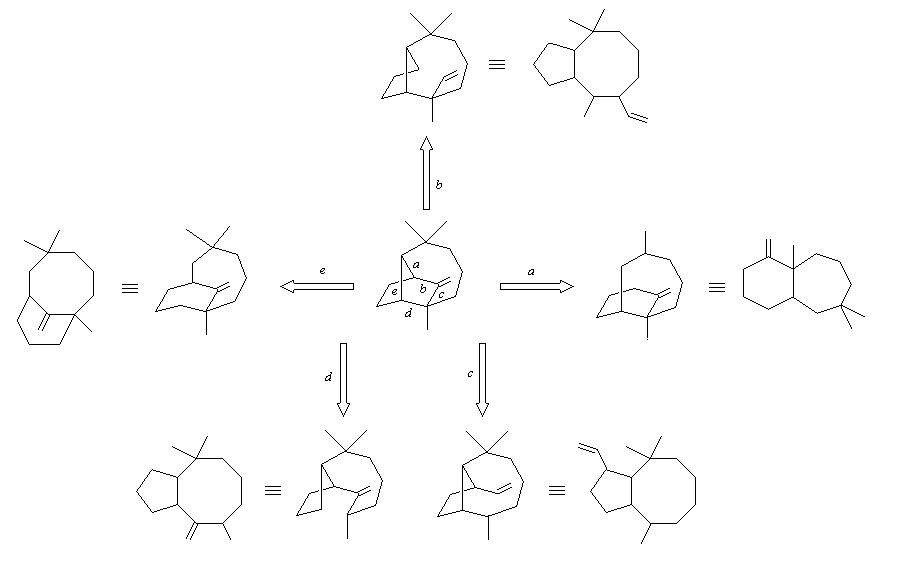
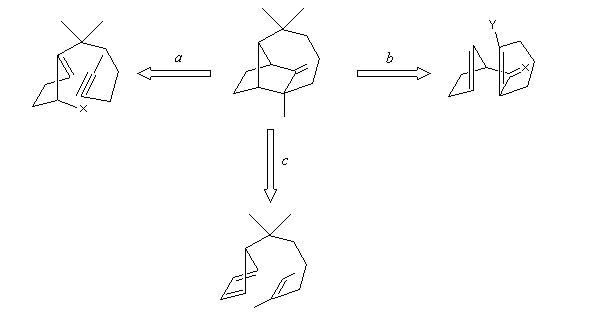
Analyse rétrosynthétique de Corey [2], [3]
Il est facile de passer de la structure de la molécule cible à la molécule A au moyen de réactions classiques.
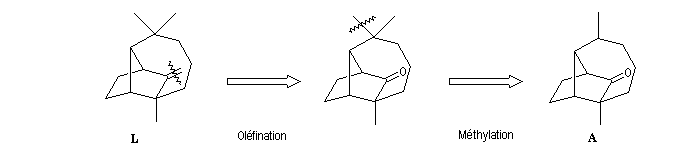
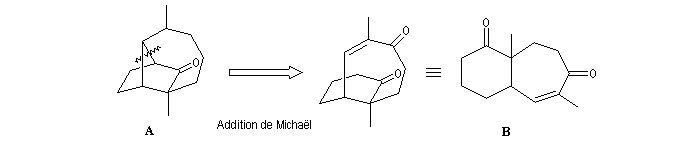
La molécule B comporte un cycle à 7 chaînons. Il est possible de l'obtenir par agrandissement de cycle en utilisant une transposition pinacolique.
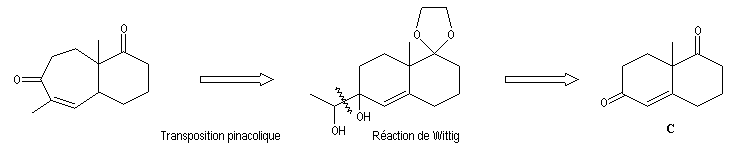
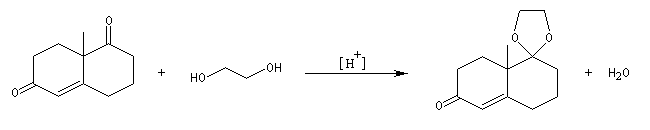
Une double liaison éthylénique exocyclique est introduit au moyen d'une réaction de Wittig.
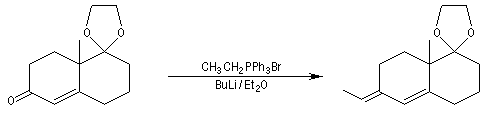
Afin de préparer la transposition pinacolique ultérieure, la double liaison subit une dihydroxylation à l'aide du tétroxyde d'osmium.
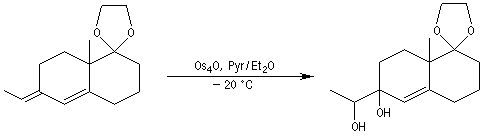
La fonction alcool secondaire réagit de façon chimiosélective avec le chlorure de tosyle. Le but de cette réaction est de préparer un tosylate afin de disposer d'un meilleur nucléofuge que le groupe OH. La protonation en milieu acide de ce groupe n'est pas possible ici car elle conduirait au déblocage du cétal.
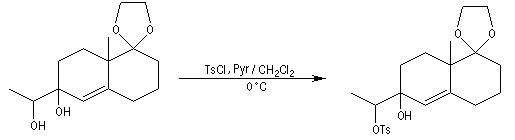
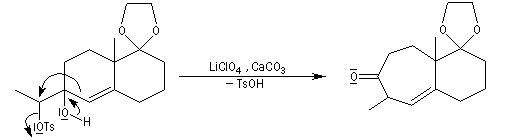
Le passage en milieu acide provoque la déprotection du groupe carbonyle et l'isomérisation de la cétone non conjuguée en a-énone, plus stable.
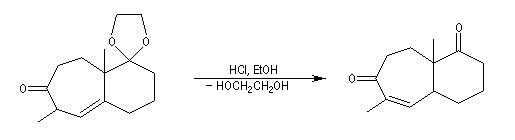
La cyclisation est réalisée par une réaction de Michaël intramoléculaire à haute température, dans un solvant à haut point d'ébullition, en présence d'une amine tertiaire comme base : la triéthylamine.
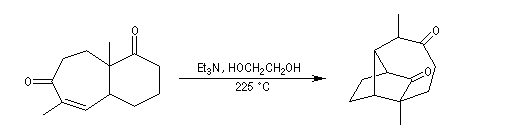
Alkylation en a du carbonyle.
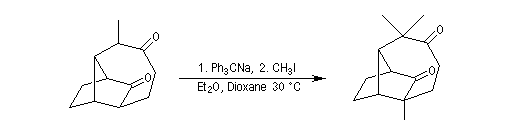
Formation chimiosélective d'un thiocétal sur le groupe carbonyle le moins encombré.
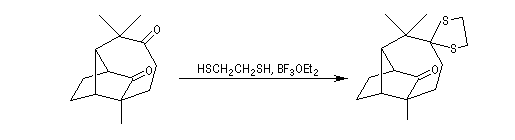
Réduction du carbonyle non protégé par LiAlH4.
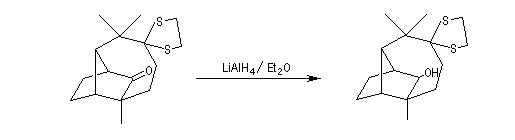
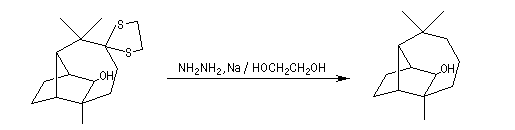
Oxydation chromique de l'alcool secondaire en cétone.
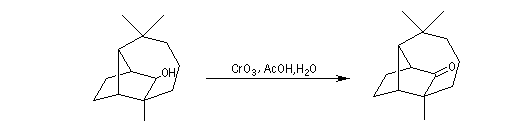
On procède donc à une méthylation par le méthyllithium suivie d'une déshydration. Noter que dans cette transformation le chlorure de thionyle est utilisé comme déshydratant électrophile.
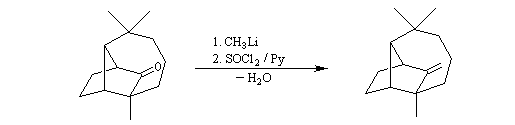
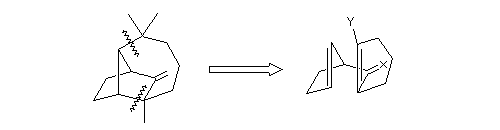
Analyse rétrosynthétique de Oppolzer
Comme on l'a dit plus haut, nous sommes ici en présence d'une stratégie comportant deux disconnections.
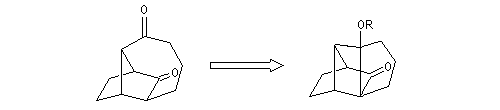
Le composé carbonylé ci-dessous est une dicétone 1,5 qui peut être obtenue par une réaction de rétrocétolisation.
Le cétol comporte un cycle de type cyclobutane qui est synthétisé grâce à une addition [2 + 2] photochimique.
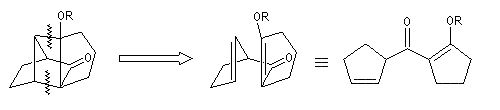
Acylation en a du carbonyle au moyen de la méthode aux énamines de Stork.
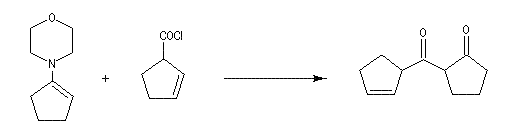
O-acylation avec formation d'un anhydride vinylique.
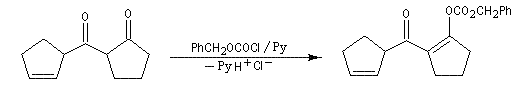
Photocyclisation intramoléculaire [2 + 2] suivie d'une rétrocétolisation. Cette suite, connue sous le nom de Réaction de de Mayo, est une méthode générale de synthèse de dicétones 1,5.
On peut la décomposer au moins formellement de la façon suivante;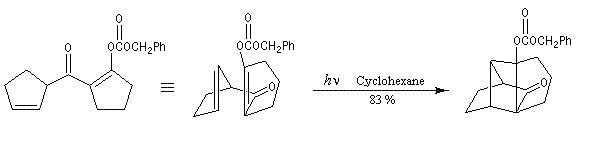
Déprotection de la fonction alcool par hydrogénolyse sur Pd/C. Cette étape conduit à un cétol instable. La réaction de rétrocétolisation qui s'effectue dans la foulée, est favorisée par l'ouverture d'un cycle contraint car comportant quatre atomes de carbone .
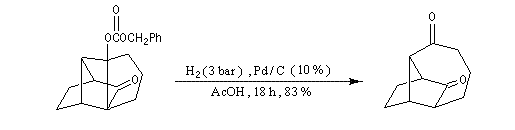
réaction de Wittig chimiosélective impliquant seulement le groupe carbonyle le moins encombré.
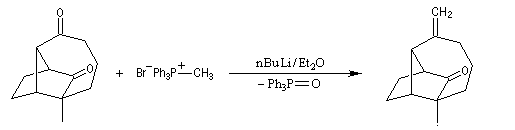
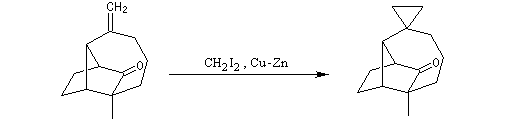
Le cyclopropane est ensuite clivé par l'hydrogène, ce qui permet d'introduire les méthyles géminés.
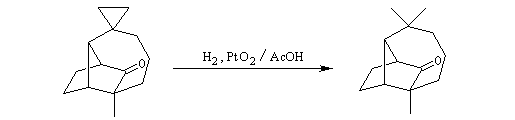
Alkylation en a du carbonyle.
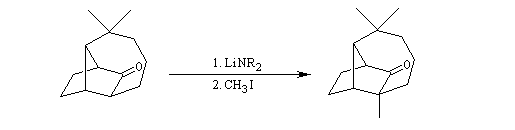
On procède donc à une méthylation par le méthyllithium suivie d'une déshydration. Noter que dans cette transformation le chlorure de thionyle est utilisé comme déshydratant électrophile.
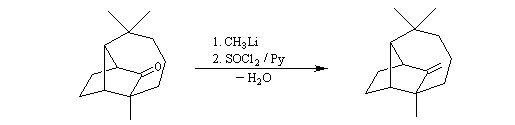
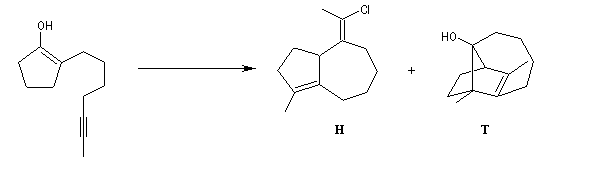
Analyse rétrosynthétique de Johnson
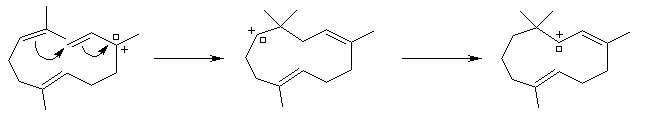
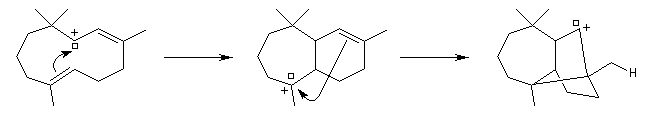
W. S. Johnson a publié en 1975 une synthèse très originale du longifolène basée sur une cascade de transpositions cationiques qui rappelent les étapes mises en jeu au cours de la biosynthèse . Au début de sa publication [5], Johnson explique comment, alors qu'il étudiait la formation d'hydroazulènes, il obtint à côté du produit attendu (H), un composé tricyclique (T),dont la structure rappelait celle du longifolène
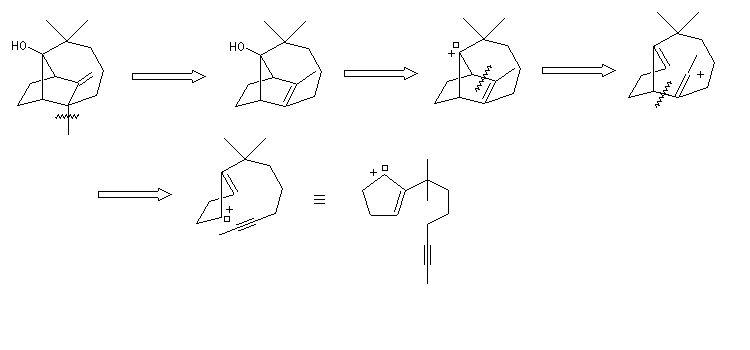
Addition en 1,4 d'un cuprate lithien sur l' a-énone. Réaction de l'éther d'énol formé avec un chlorure d'acyle.
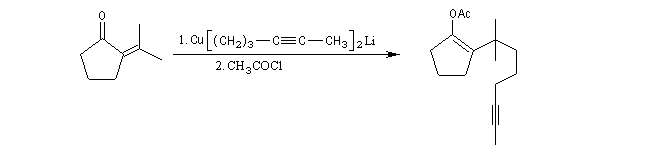
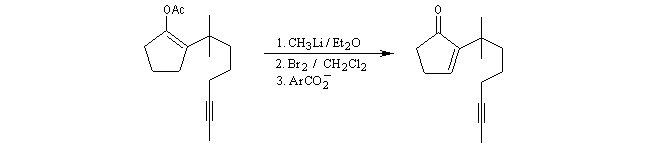
Réduction du carbonyle par LiAlH4.
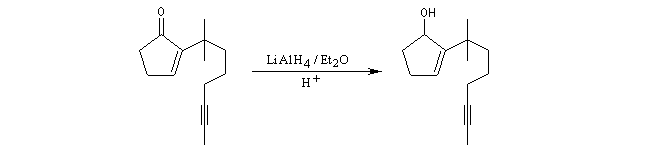
Formation d'un carbocation à partir de l'alcool par action de l'acide trifluoroacétique.
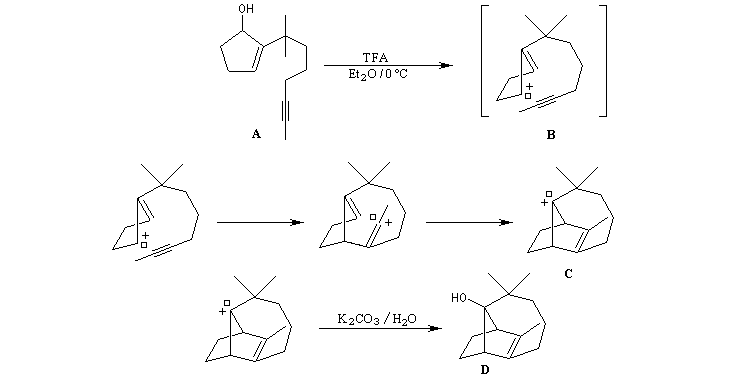
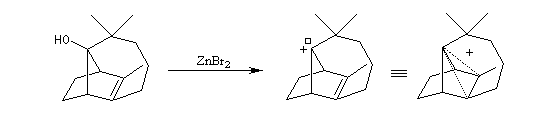
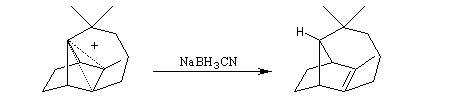
Isomérisation de la double liaison en milieu acide.
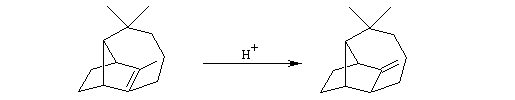
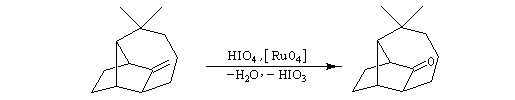
Alkylation en a du carbonyle.
On procède donc à une méthylation par le méthyllithium suivie d'une déshydration. Noter que dans cette transformation le chlorure de thionyle est utilisé comme déshydratant électrophile.
Applications
Le longifolène est utilisé comme composant de parfums.
La présence d'une double liaison permet la préparation aisée de dérivés organoboriques chiraux utilisés en synthèse énantiosélective [9].
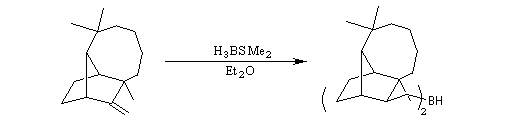
Références
[1] P. Naffa, G. Ourisson, Bulletin de la Société chimique de France, (1954), 1410.
[2] Corey, E. J. et al. J. Am. Chem. Soc. 1961, 83, 1251.
[3] Corey, E. J. et al. J. Am. Chem. Soc. 1964, 86, 478.
[4] Oppolzer, W.; Godel, T. J. Am. Chem. Soc. 1978, 100, 2584.
[5] Volkman, Andrews, Johnson, J. Am. Chem. Soc. 1975, 97, 4777.
[6] Gregory A. Ho, Dustin H. Nouri, and Dean J. Tantillo*
Department of Chemistry, University of California, Davis, One Shields Avenue, Davis, California 95616 J. Org. Chem., 2005, 70 (13), pp 5139–5143
[7] de Mayo, P. Takeshita, H. Sattar, A. B. M. A. Proc. Chem. Soc., London, 1962, 119.
[8] F. A Carey, R. J Sundberg Advanced Organic Chemistry (Plenum Press 1990).
[9] Jadhav, P. K.; Brown H. C. J. Org. Chem. 1981, 46, 2988.
.
Links
Conférence Nobel de E. J. Corey
de Mayo reaction
For educational purpose - No commercial use.
Page created by Gérard Dupuis - Lycée Faidherbe LILLE.
gdupuis-prof@faidherbe.org
This site was last updated June 2010.