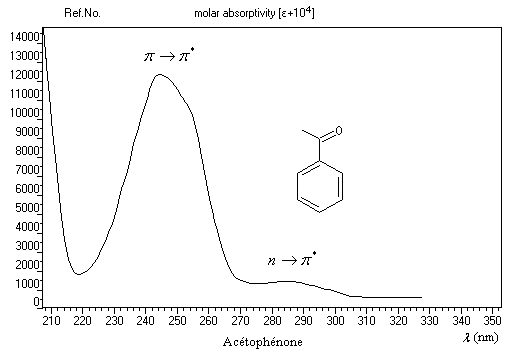
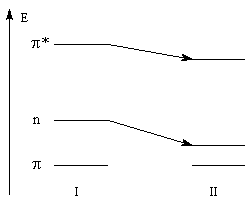
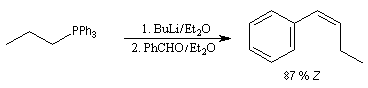
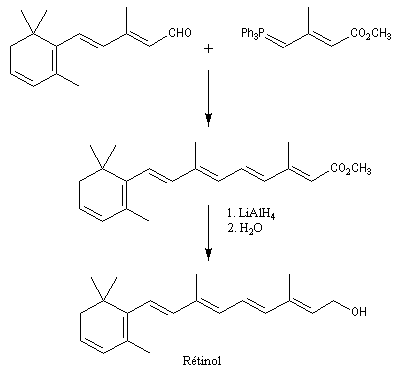
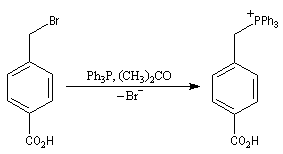
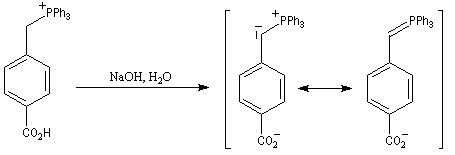
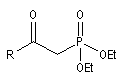
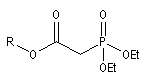
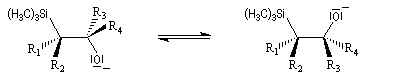
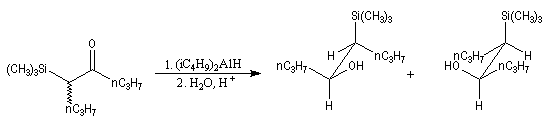
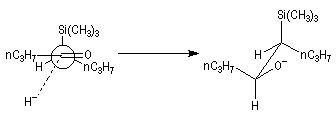
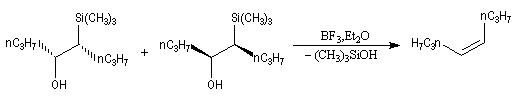
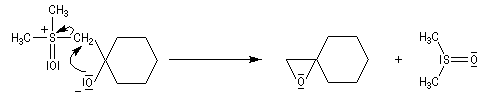
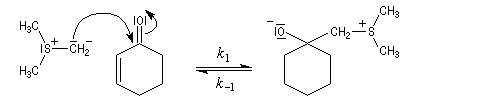
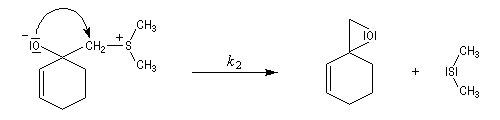
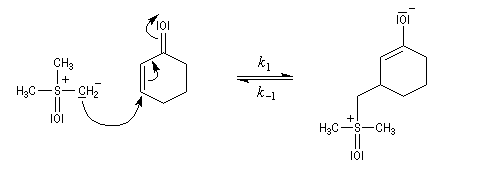
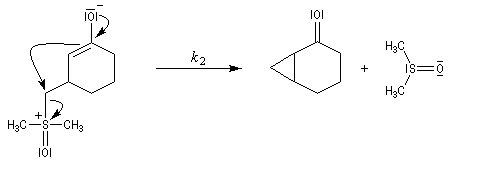
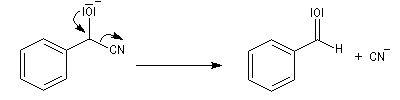
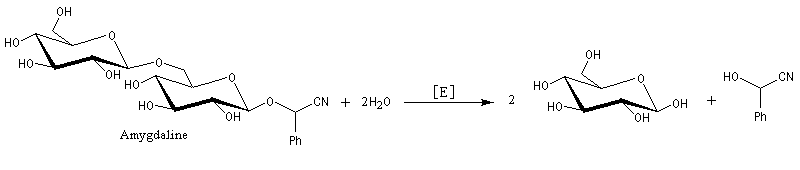
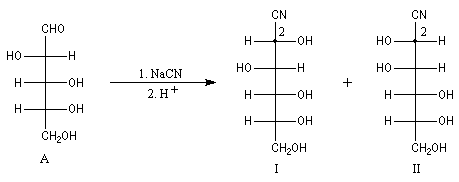
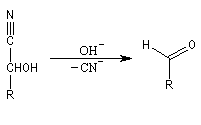
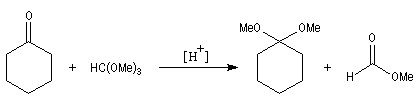
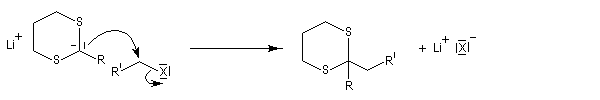
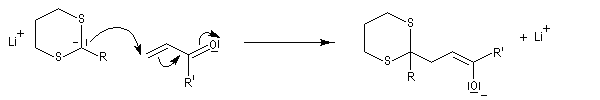
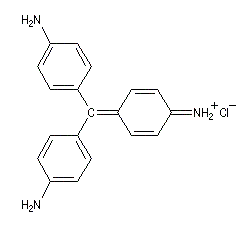
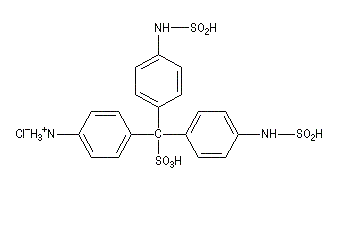
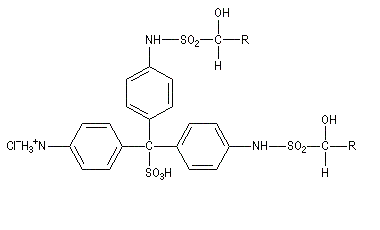
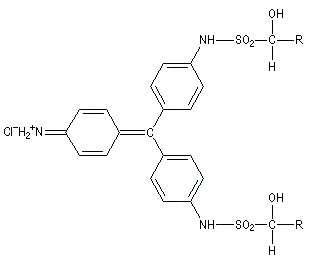
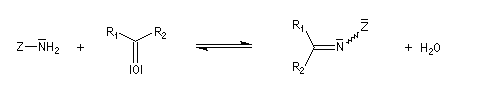
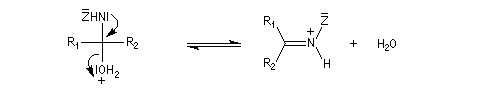
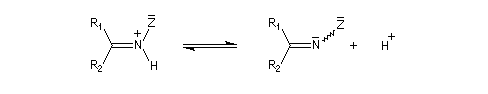
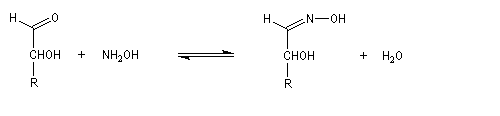
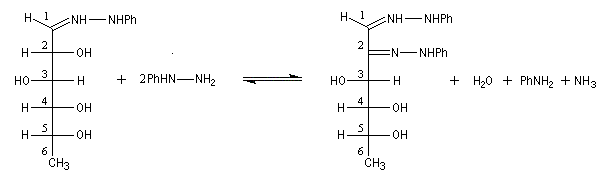
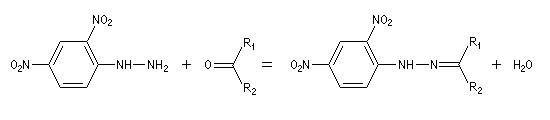
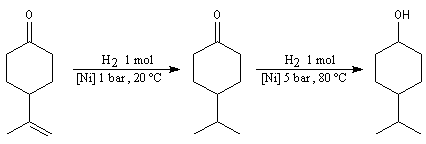
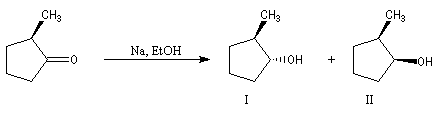
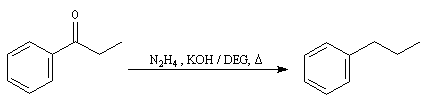
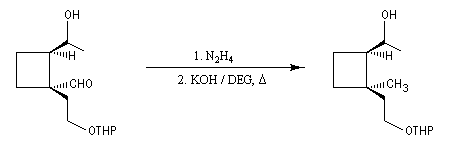
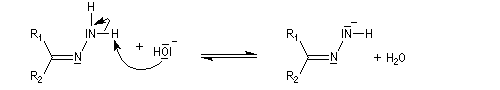
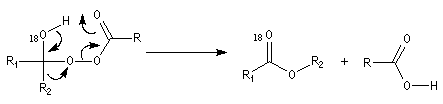
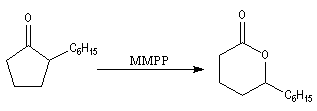
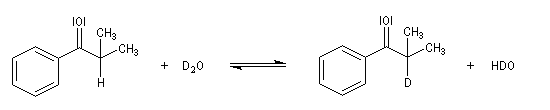
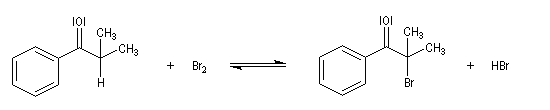
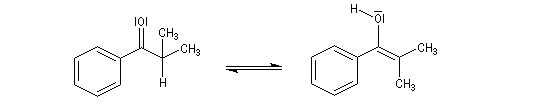
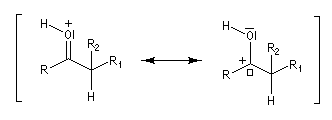
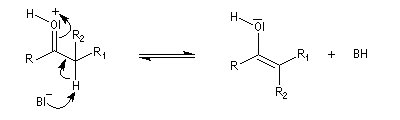
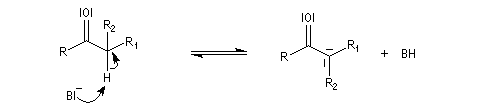
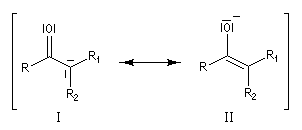
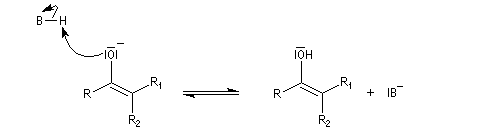
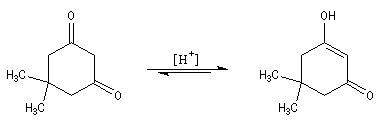
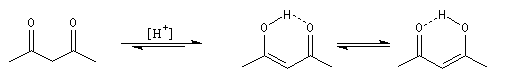
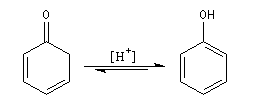
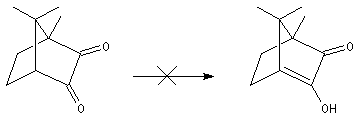
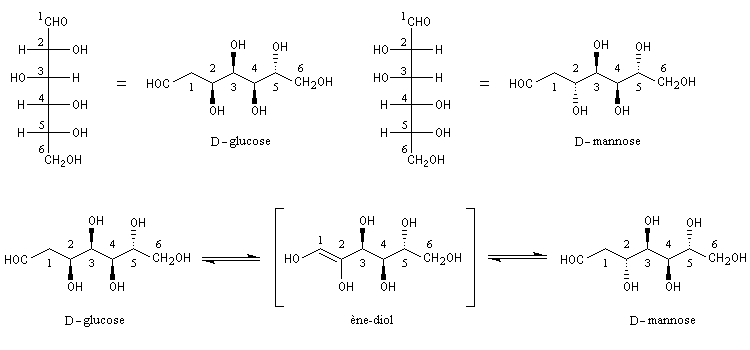
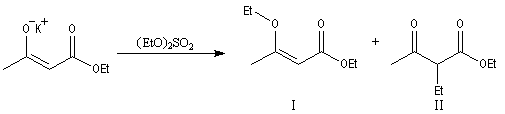
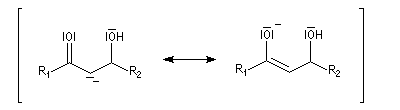
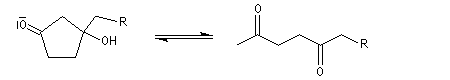
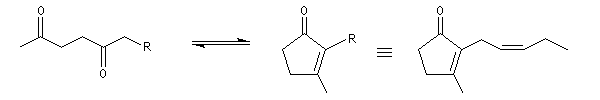
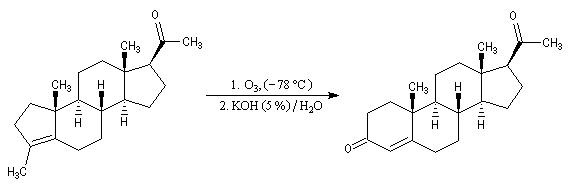
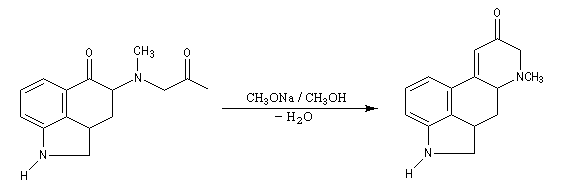
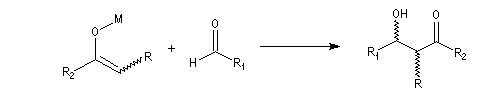
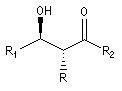
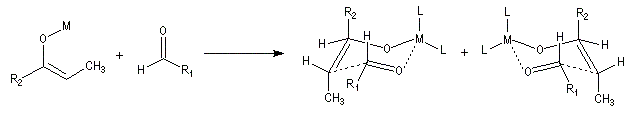
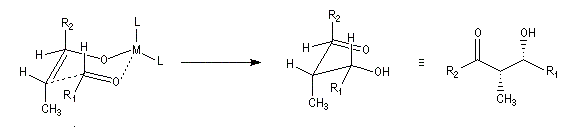
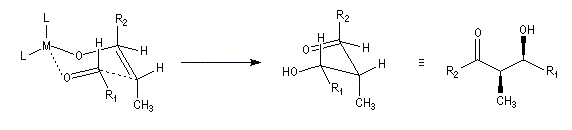
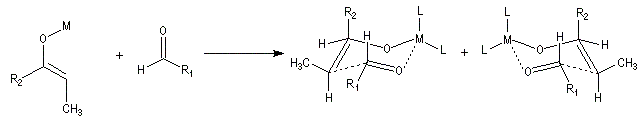
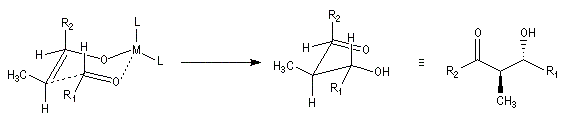
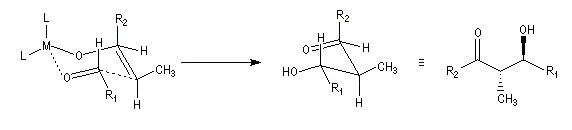
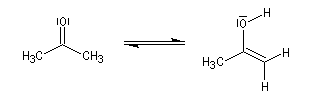
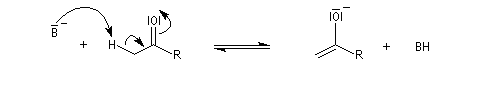
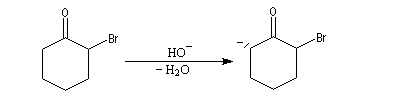
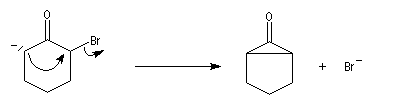
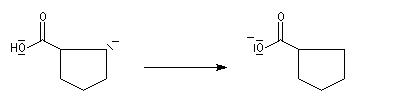
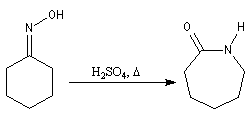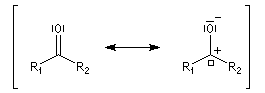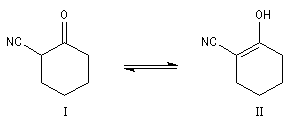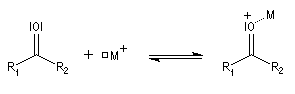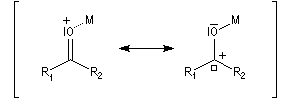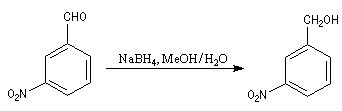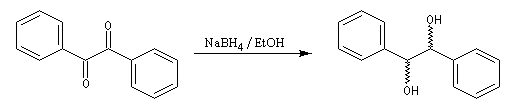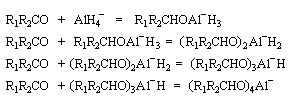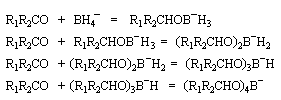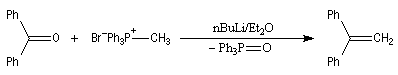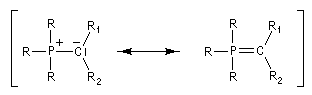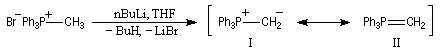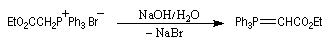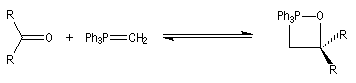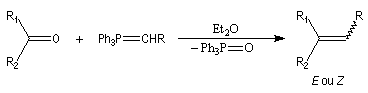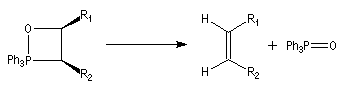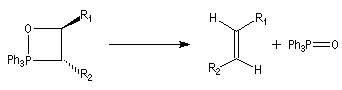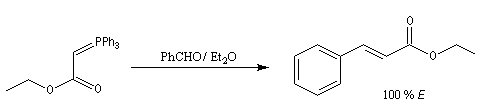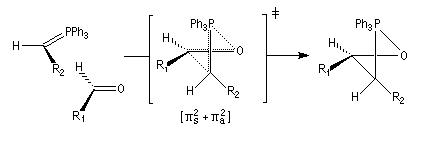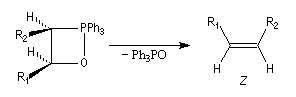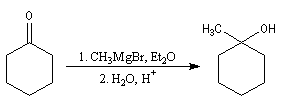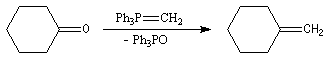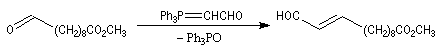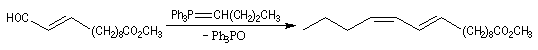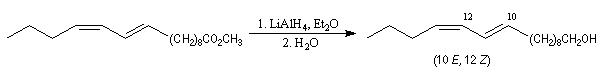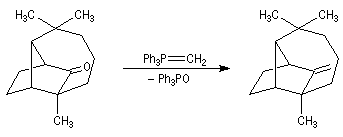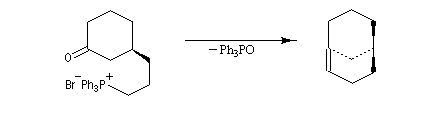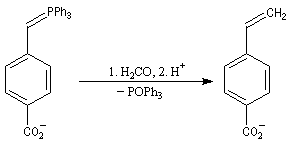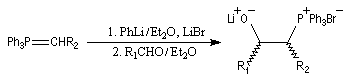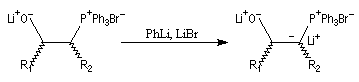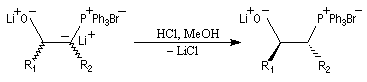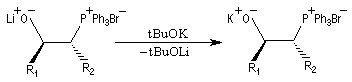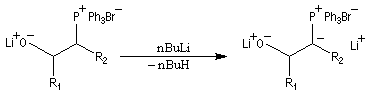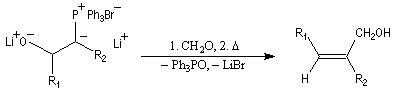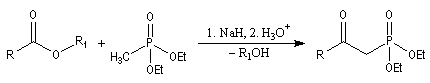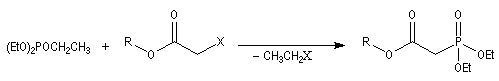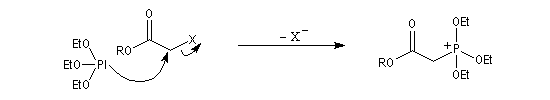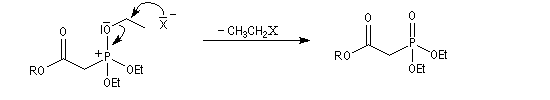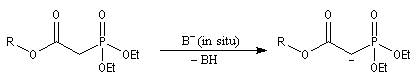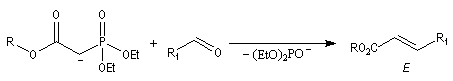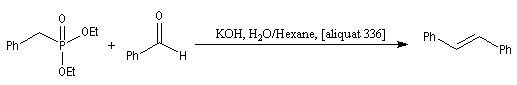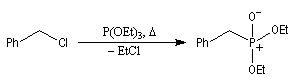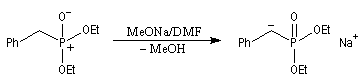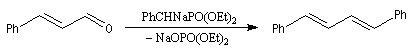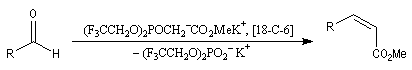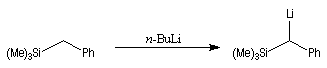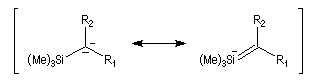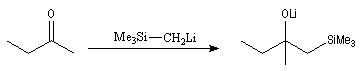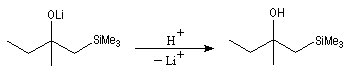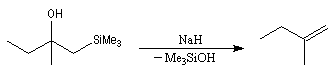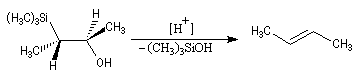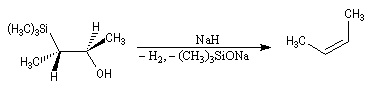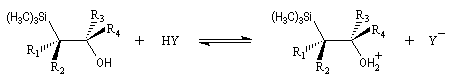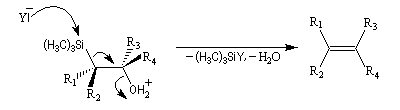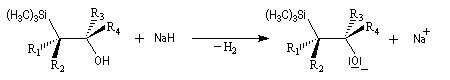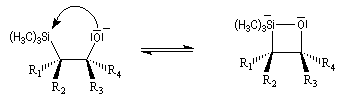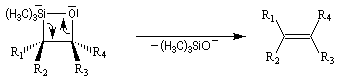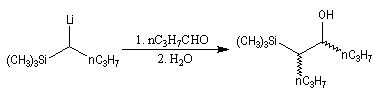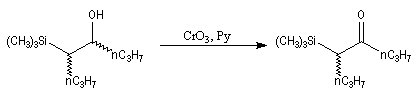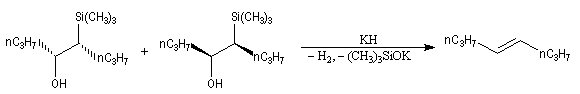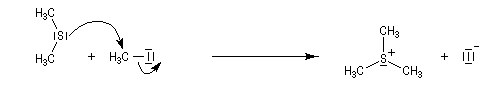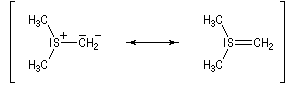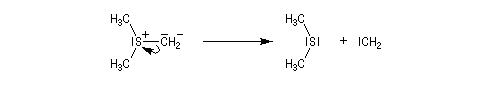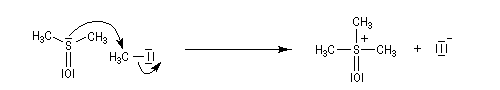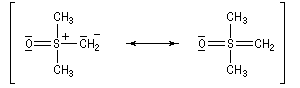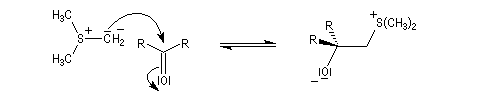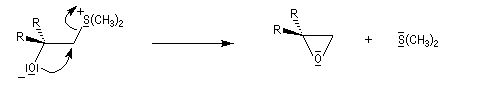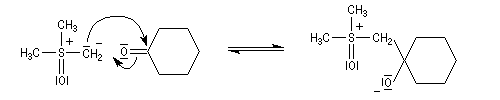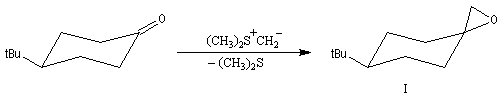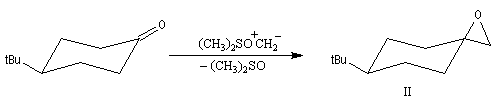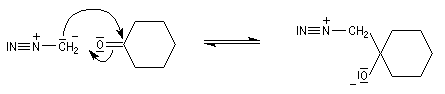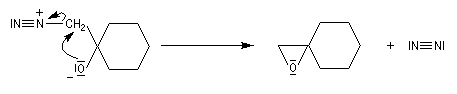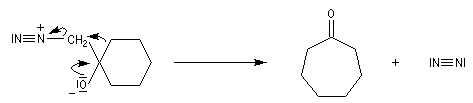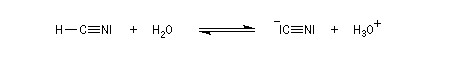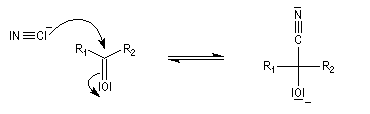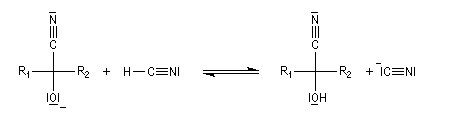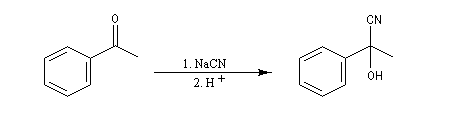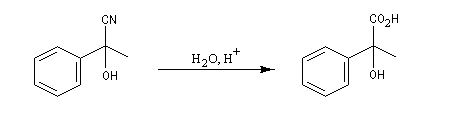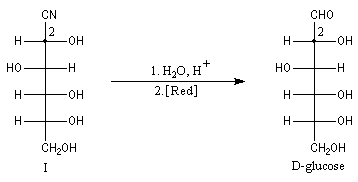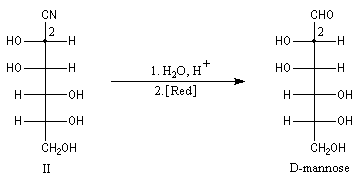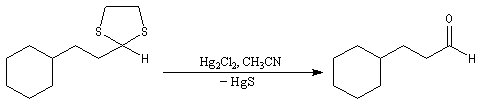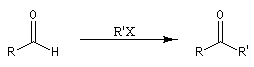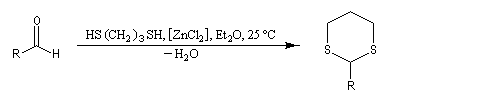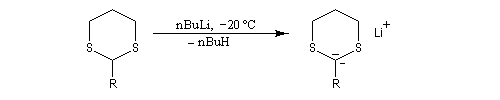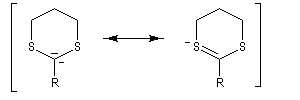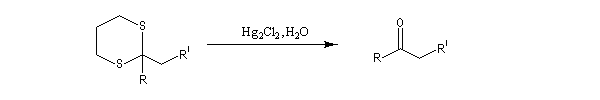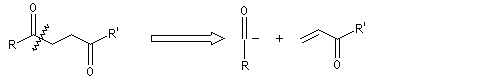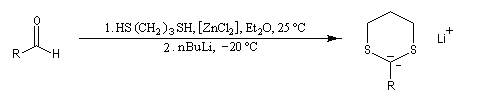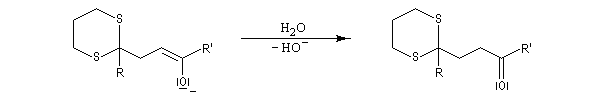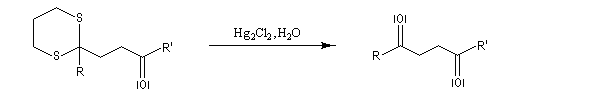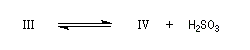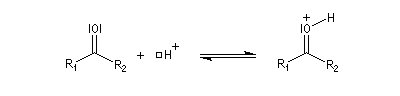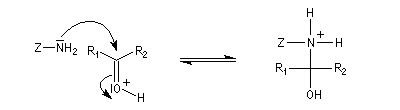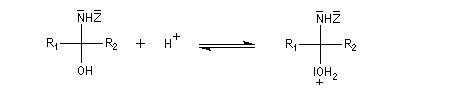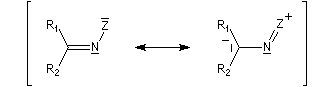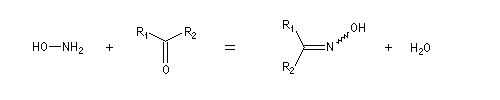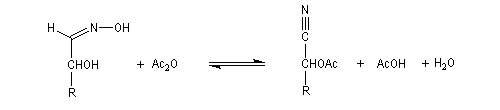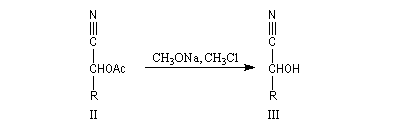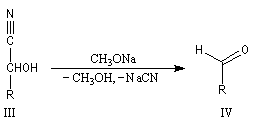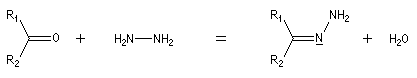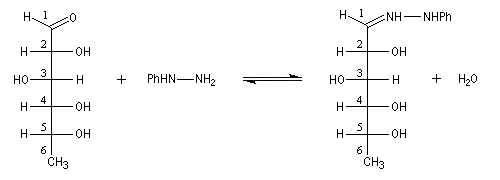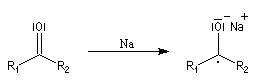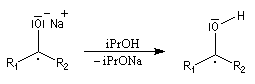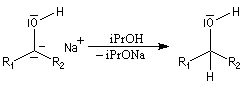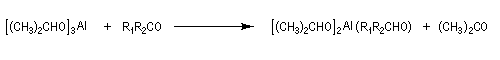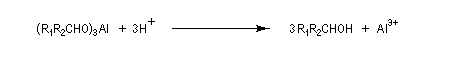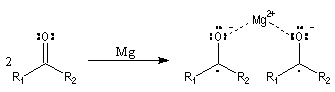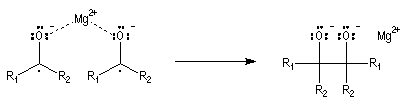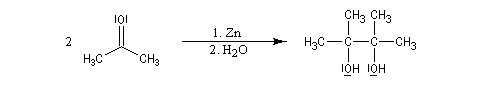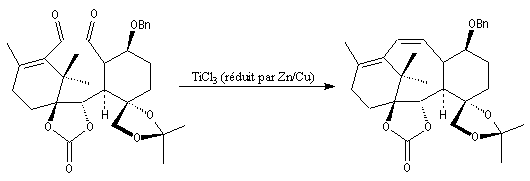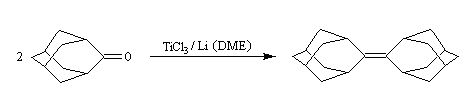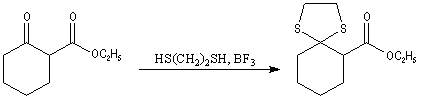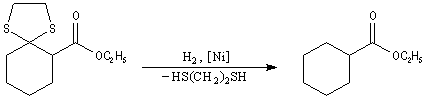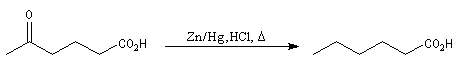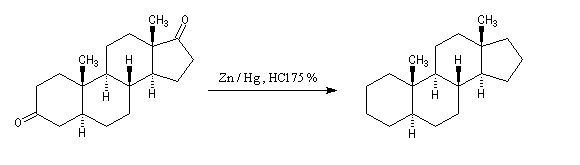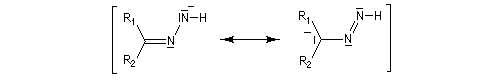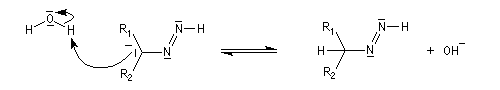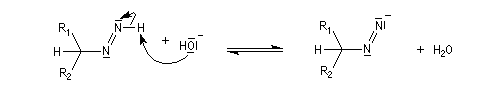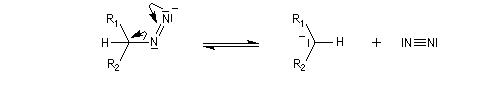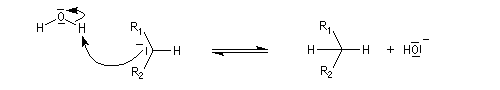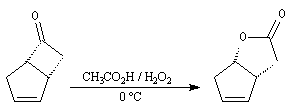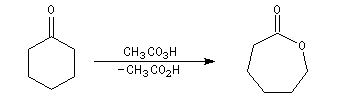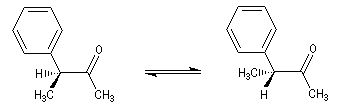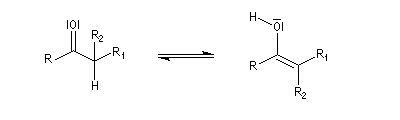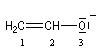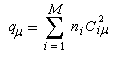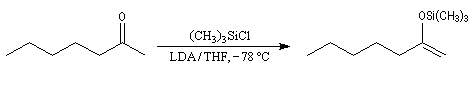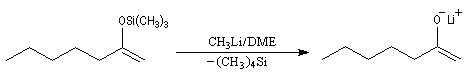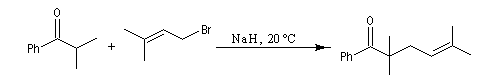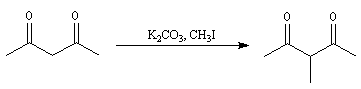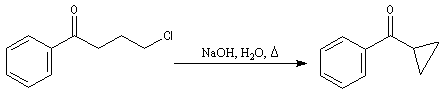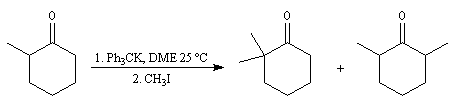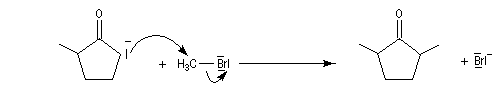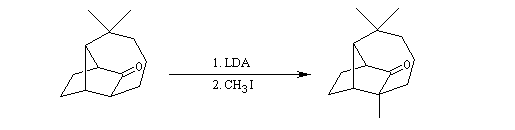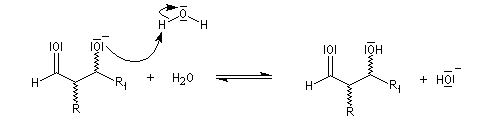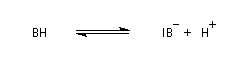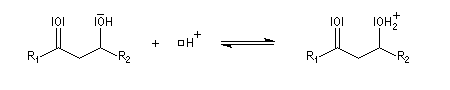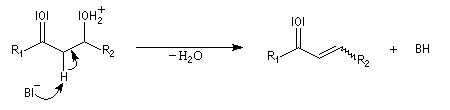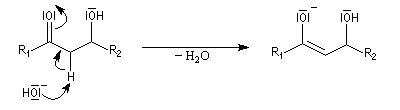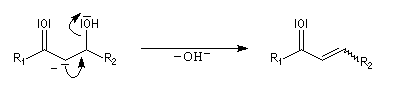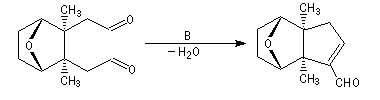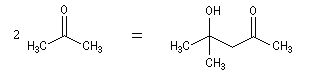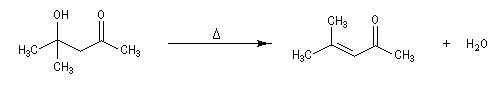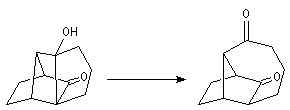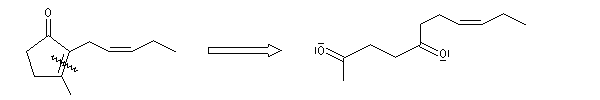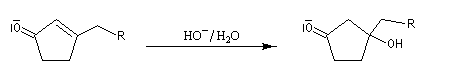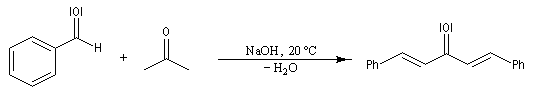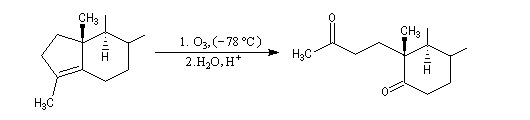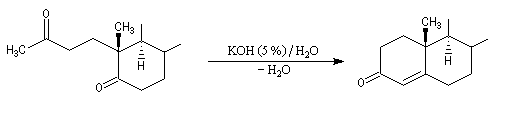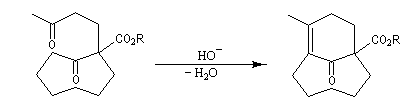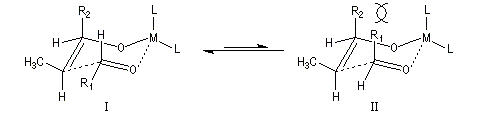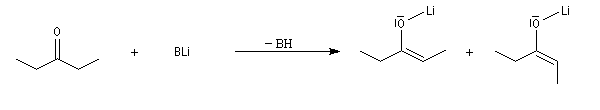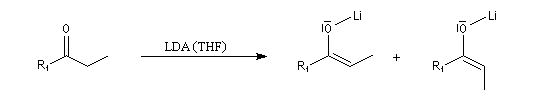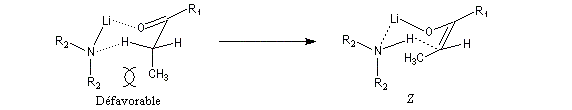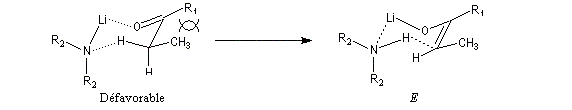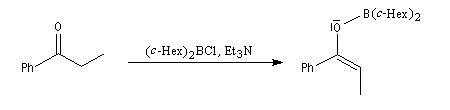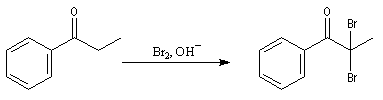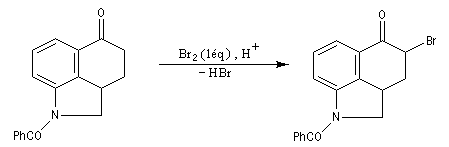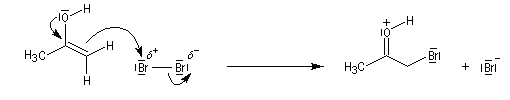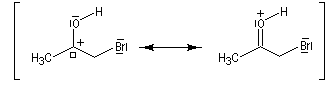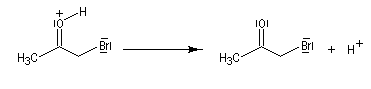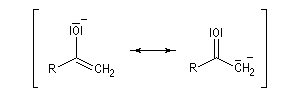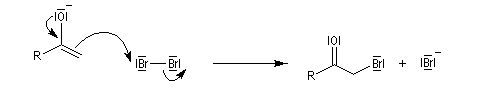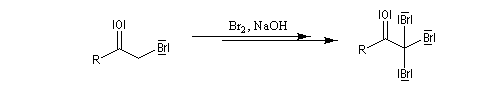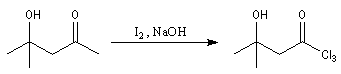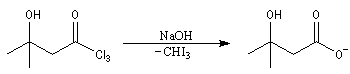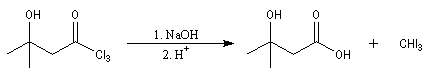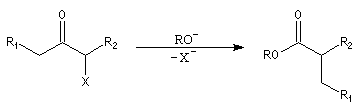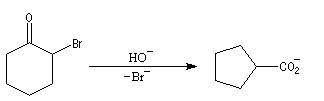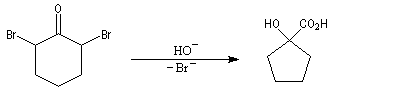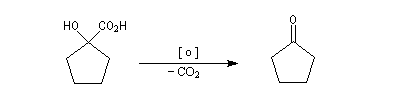Du fait de leur importance, les principales propriétés physiques, et chimiques des a-énones sont étudiées dans un chapitre particulier de la page consacrée aux systèmes conjugués.
Carbonylés importants
|
|
Le méthanal est préparé par oxydation du méthanol.
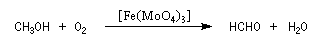
Il est utilisé notamment dans la synthèse de la bakélite.
|
|
|
L'éthanal préparé actuellement par oxydation de l'éthène grâce au procédé Wacker.
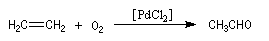
Un ancien procédé consistait à hydrater l'éthyne (acétylène).
|
|
|
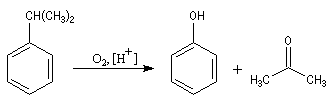
La propanone est préparée à partir du propène par le procédé Wacker. C'est aussi un sous-produit de la synthèse du phénol par le procédé Hock dont le bilan
revient à oxyder le cumène par l'oxygène de l'air.
|
Plusieurs composés carbonylés sont utilisés comme composants de parfums. Le célèbre parfum N° 5 de la maison Chanel doit sa note de tête caractéristique à la présence d'un aldéhyde dont la chaîne la plus longue comporte 11 atomes de carbone : le 1-méthylundécanal [66].
Nomenclature
Aldéhydes
On distingue les séries acyclique et cyclique.
- Série acyclique
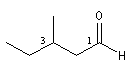
Le nom est formé en ajoutant le suffixe al au nom de l'hydrocarbure correspondant à la chaîne principale. L'atome de carbone du groupe porte le numéro 1 qui n'est pas mentionné.
|

|
|
|
hexanal |
3-méthylpentanal |
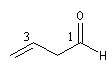
Pour les composés polyfonctionnels on utilise les suffixes dial et trial. Si le groupe CHO est prioritaire dans un composé polyfonctionnel, il porte le numéro 1.
|

|
|
|
hexanedial |
but-3-énal |
- Série cyclique
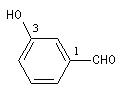
Lorsque le groupe CHO est fixé sur un atome de carbone faisant partie d'un système non aromatique on ajoute la terminaison
carbaldéhyde au nom du système cyclique.
Lorsque le groupe CHO est fixé sur un cycle benzénique, le composé est nommé comme dérivé du benzaldéhyde.
|

|
|
|
cyclohexanecarbaldéhyde |
3-hydroxybenzaldéhyde |
Cétones
Comme dans le cas précédent, on distingue les séries cyclique et acyclique.
|
|
|
|
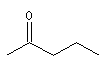
pentan-2-one |
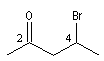
4-bromopentan-2-one |
Dans le cas où le groupe carbonyle n'appartient pas à la chaîne principale. Le substituant H3C-CO- est désigné par acétyle.
|
|
|
|
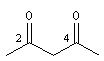
pentane-2, 4-dione |
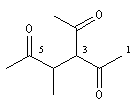
3-acétyl-4-méthylhexane-2, 5-dione |
|
|
|
|
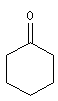
cyclohexanone |
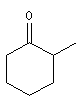
2-méthylcyclohexanone |
Le nom peut être formé en nomenclature radico-fonctionnelle par assemblage. On utilise l'ordre alphabétique
pour énoncer les groupes qui sont précédés éventuellement de di, tri etc.
|
|
|
|
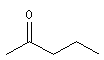
méthylpropylcétone |
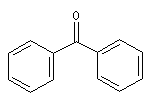
diphénylcétone |
Les composés dans lesquels la fonction aldéhyde ou la fonction cétone ne sont pas
prioritaires sont respectivement nommés en utilisant les préfixes formyle
et oxo.
|
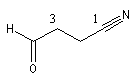
|
|
|
3-formylpropanenitrile |
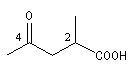
acide 2-méthyl-4-oxopentanoïque |
Propriétés physiques
Constantes physiques
Voici quelques résultats expérimentaux concernant le moment dipolaire et les énergies de liaisons
de quelques composés carbonylés.
|
Molécule |
HCHO |
CH3CHO |
CH3COCH3 |
|
p (D) |
2,27 |
2,73 |
2,84 |
|
Liaison |
C-C |
C-O |
C=C |
C=O |
|
D (kJ.mol-1) |
347 |
360 |
610 |
740 |
Ces valeurs appellent deux remarques :
- on note une valeur élevée du moment dipolaire des molécules qui traduit la polarité
importante du groupe carbonyle ;
- l'énergie de dissociation particulièrement élevée de la double liaison CO qui est supérieure au double de
celle d'une liaison simple entre ces atomes.
Températures de changement d'état
|
Composé |
TF (°C) |
TE (°C) |
d |
|
HCHO |
- 118 |
19 |
- |
|
CH3CHO |
- 123 |
21 |
0,79 |
|
CH3CH2CHO |
- 81 |
48 |
0,80 |
|
CH3COCH3 |
- 95 |
56 |
0,79 |
Les températures de changement d'état sont plus élevées que pour les alcanes mais moins élevées que pour les alcools de même masse molaire.
Elle traduisent une association des molécules par des forces de type dipôle-dipôle.
Structure électronique du groupe carbonyle
Méthode des orbitales moléculaires
Le groupe carbonyle peut être décrit par deux méthodes qui sont souvent utilisées conjointement et qui se complètent de façon utile. La méthode des orbitales moléculaires et celle, plus fruste, de la mésomérie.
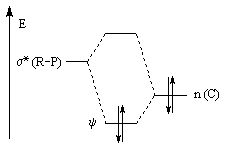
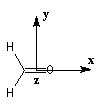
Nous donnons ci-dessous un diagramme qualitatif des orbitales moléculaires pour une molécule de méthanal. Les orbitales moléculaires des autres composés carbonylés peuvent être obtenues par la méthode des perturbations.
|
|
Les expériences de diffraction montrent que les atomes de carbone, d'oxygène et d'hydrogène sont situés dans un même plan. La molécule
présente deux plans de symétrie :
- le plan contenant tous les atomes (xOy) ;
- le plan passant par les atomes de carbone et d'oxygène et bissectant l'angle entre les liaisons C-H (xOz).
|
Les orbitales atomiques impliquées peuvent être classées en deux catégories. Celles qui sont symétriques par réflexion par rapport au plan de la molécule
et celles qui sont antisymétriques par réflexion par rapport à ce plan.
La combinaison de ces orbitales conduit à un système de 6 orbitales moléculaires dont seulement trois sont représentées.
|
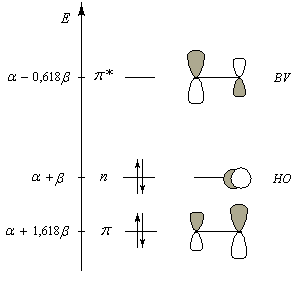
|
Le diagramme d'orbitales moléculaires représenté à gauche regroupe les OM p liante, n non liante et p* antiliante.
Les orbitales frontières sont :
- HO : orbitale n, qui correspond à peu de chose près à une OA p(y) de l'atome d'oxygène ;
- BV : orbitale p* avec un gros coefficient sur l'atome de carbone.
|
L'expression mathématique des Spectroscopie infrarouge
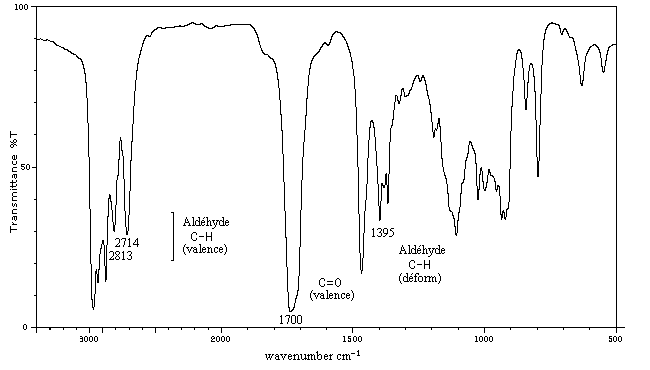
C'est une méthode de choix pour l'étude des composés carbonylés. Le nombre d'onde de la vibration de valence du groupe
carbonyle dépend du type de composé mais apparaît nettement sous la forme d'une bande intense dans une région assez dégagée du spectre.
L'exemple ci-dessous concerne la 4-méthylpentan-2-one.
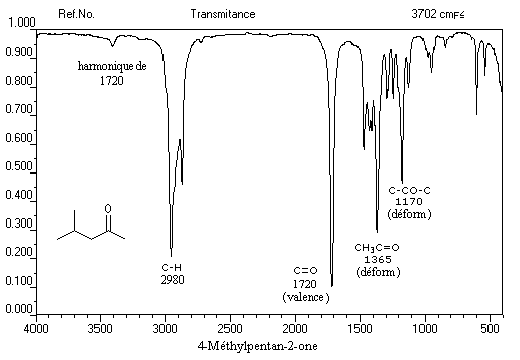
Les aldéhydes possèdent en outre une absorption caractéristique dûe à la vibration de valence de la liaison C-H. Elle se présente sous la forme d'un pic fin.
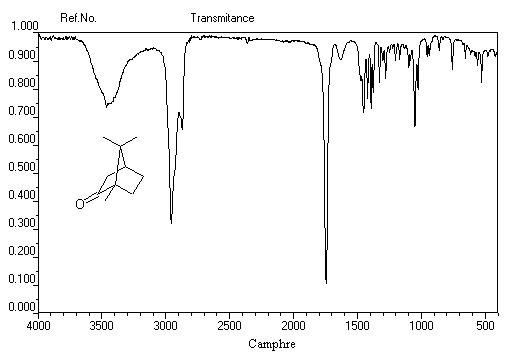
Notons deux propriétés importantes concernant le nombre d'onde de la vibration d'élongation du carbonyle. On observe une augmentation du nombre d'onde dans le cas de cétones cycliques lorsque la taille du cycle diminue.
L'exemple suivant concerne la pentan-3-one.
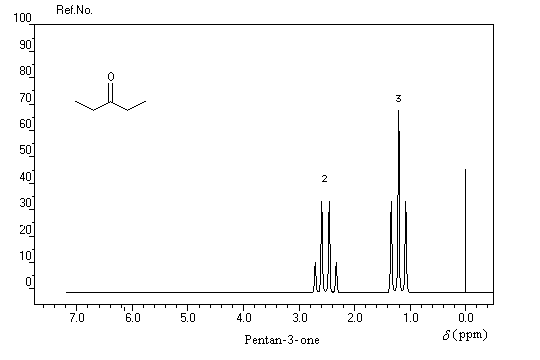
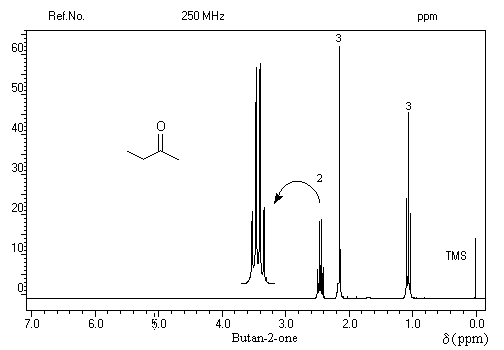
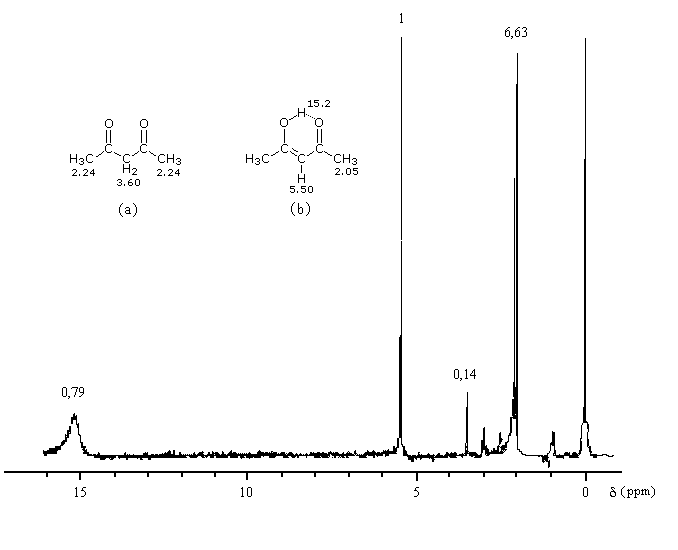
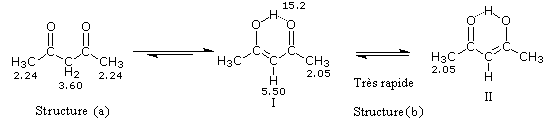
On repère facilement les méthylcétones par la présence d'un pic vers 2 ppm comme dans l'exemple ci-dessous.
Elle est interdite de symétrie ce qui explique une intensité relativement faible.
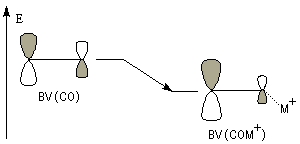
L'association entre l'atome d'oxygène et M+, draine les électrons vers M+. L'ensemble
(COM+) se comporte comme un groupe plus électronégatif que l'oxygène seul. Ce groupe possède une énergie plus basse. Il en résulte un abaissement
de l'énergie de la BV et une augmentation du coefficient au niveau de l'atome de carbone de cette orbitale.
Le phénomène que nous venons de décrire, porte le nom d'assistance électrophile.
Géométrie de l'addition nucléophile
Commençons par examiner la direction d'attaque du nucléophile dans le plan perpendiculaire au plan du carbonyle.
Avec les nucléophiles peu encombrés, la géométrie de l'addition est gouvernée par le recouvrement
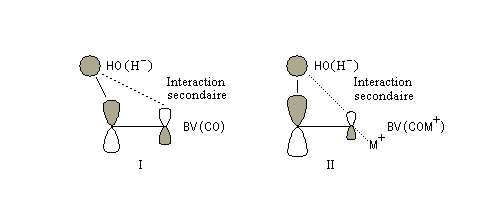
des orbitales frontières des réactifs. Raisonnons dans le cas de l'addition d'un ion hydrure. Ce dernier
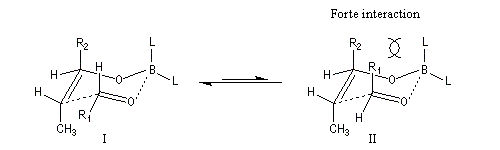
intervient par sa plus haute om occupée (1 s). Le carbonyle intervient par la plus basse om vacante. Le schéma ci-dessous résume la situation en l'absence (I) puis en présence
d'une activation électrophile (II).
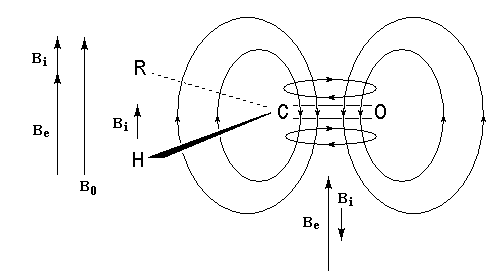
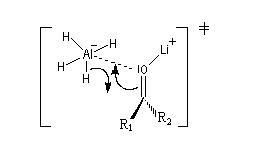
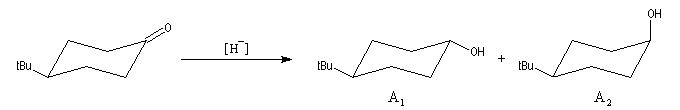
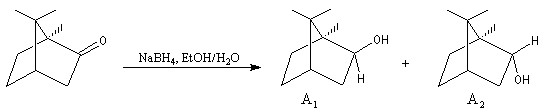
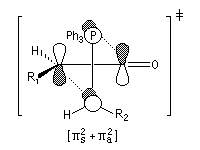
Le phénomène a été étudié dans les années 70 par H. B. Bürgi, J. D. Dunitz et J. M. Lehn [43]. L'angle d'attaque, appelé usuellement angle de Bürgi-Dunitz, est voisin de 105° (figure I). Ce résultat s'interprète en considérant le recouvrement des orbitales. La géométrie de l'interaction maximise l'interaction stabilisante tandis qu'elle minimise l'interaction secondaire déstabilisante entre les lobes orbitalaires de signes contraires (cette interaction est figurée en pointillés sur la figure.)
En présence d'activation électrophile par l'acide de Lewis M+, le coefficient sur l'atome de carbone augmente ce qui a pour conséquence d'augmenter l'intensité de l'interaction prinicpale stabilisante et de diminuer l'interaction secondaire déstabilisante. L'angle d'attaque diminue et passe à une valeur voisine de 95° (figure II.)
L'exemple suivant constitue une vérification expérimentale de ce qui précède. La molécule présente deux groupes carbonyles. Celui qui est réduit préférentiellement est celui qui est adjacent
à l'atome de carbone le plus encombré.
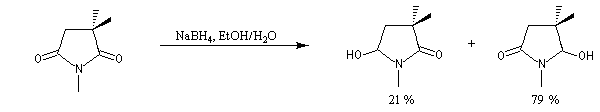
Ce résultat en apparence paradoxal s'explique par le fait que la trajectoire suivie par le nucléophile n'est pas perpendiculaire au plan du groupe carbonyle.
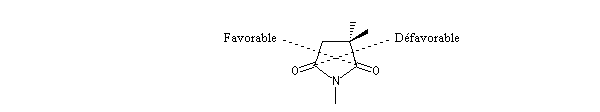
La trajectoire de Bürgi-Dunitz intervient dans le modèle de modèle de Felkin-Anh étudié dans le chapitre de stéréochimie dynamique.
Remarque : l'angle entre la direction d'attaque du nucléophile et celui du carbonyle dans le plan joue également un rôle dans le cas où l'on a affaire à des aldéhydes possèdant un atome de carbone chiral en a du carbonyle (angle de Flippin-Lodge). Cet effet est étudié dans le chapitre consacré à la stéréochimie dynamique.
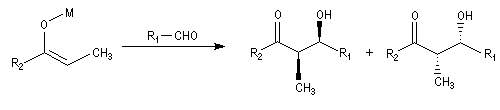
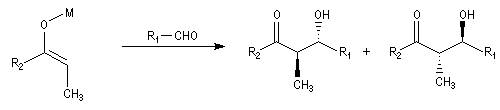
Cas où les faces du carbonyle sont prochirales
Les deux faces d'un groupe carbonyle lié à deux substituants différents sont prochirales et ces faces sont nommées en utilisant la convention de Hanson. L'exemple ci-dessous
concerne la molécule d'éthanal.
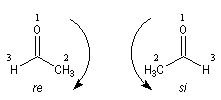
Envisageons l'addition d'un réactif A - B achiral sur le groupe carbonyle. Si R1 et R2 sont différents,
on obtient a priori deux produits selon que l'addition implique l'une ou l'autre des deux faces du groupe carbonyle.
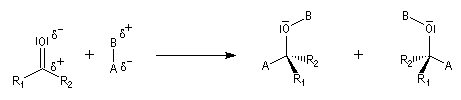
Deux cas peuvent se présenter :
Lorsqu'elle s'accompagne de l'apparition de protons diastéréotopiques, l'addition d'un nucléophile sur les faces prochirales d'un groupe carbonyle peut être aisément détectée en spectroscopie de RMN.
Présence d'un carbone asymétrique en a du groupe carbonyle
La présence d'un centre chiral en a du groupe carbonyle pose un problème d'induction asymétrique, la règle de Cram ainsi que les modèles de Felkin et de Felkin-Anh sont examinés dans le chapitre consacré à la stéréochimie dynamique.
Addition nucléophile des hydrures
Réactifs donneurs d'hydrure
L'ion hydrure contient l'élément hydrogène au degré (-I). C'est donc un réducteur potentiel de fonctions insaturées.
Les hydrures alcalins LiH, NaH, KH sont connus depuis longtemps. Cependant ces composés ne sont guère utilisables comme réducteurs car ce sont des bases très fortes et l'ion H- n'y manifeste pas
de propriétés nucléophiles. En revanche dans les complexes d'hydrures d'aluminium ou de bore, l'hydrogène est lié par une liaison covalente polarisée. Ce sont des réactifs gros et polarisables, permettent de transférer l'ion H- vers un substrat insaturé en exaltant sa capacité nucléophile.
Le tétrahydruroaluminate de lithium LiAlH4 (en anglais : lithium alumino hydride LAH) est un composé ionique qui se présente sous la forme d'un solide blanc, stable dans l'air sec mais extrêmement réactif. On le conserve généralement dans une huile minérale.
Il a été préparé en 1947 par H. J. Schlesinger. On l'utilise dans l'éthoxyéthane ou le THF. Il est absolument incompatible avec l'eau qu'il réduit avec explosion à cause du dégagement d'hydrogène et du caractère très exothermique de la réaction. On peut le préparer par la réaction suivante :
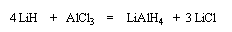
LiAlH4 réduit de nombreuses doubles liaisons polarisées. Il est peu sélectif.
|
|
Le tétrahydroborate de sodium NaBH4 (on utilise hydro et non hydruro pour désigner le ligand de l'ion complexe en chimie du bore pour des raisons historiques.) a été préparé par H. I. Schlesinger et H. C. Brown en 1943 dans le cadre du "projet Manhattan". On peut l'obtenir en faisant réagir l'hydrure de sodium et le diborane.
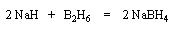
|
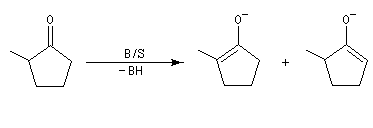
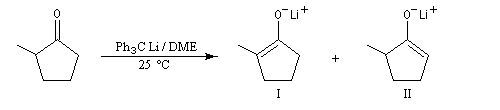
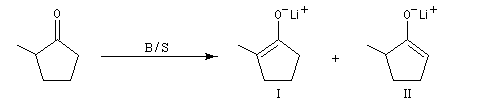
NaBH4 est beaucoup moins réactif que LiAlH4 (la liaison B-H est plus forte que la liaison Al -H). Il offre de ce fait, l'avantage d'une bien plus grande sélectivité. Pour les composés carbonylés, l'ordre de réactivité est le suivant : a-énones < cétones < a-énals (aldéhydes conjugués) <
aldéhydes.
En général, un certain type de groupement carbonyle peut être réduit sélectivement en présence d'un
autre groupe carbonyle d'une catégorie moins réactive.
On réalise ces réactions en utilisant un excès de tétrahydroborate de sodium, dans des mélanges de solvants tels que méthanol ou d'éthanol avec du dichlorométhane. A la différence de LiAlH4, il n'est pas totalement incompatible avec l'eau dans laquelle il se dissout assez bien. Il réagit cependant lentement avec l'eau pour donner du dihydrogène. C'est la raison pour laquelle il faut l'utiliser en excès en présence d'un milieu aqueux.
Notons que ce réactif réduit les chlorures d'acyles en alcools.
Dans l'exemple suivant, le groupe nitro du substrat n'est pas altéré alors qu'il serait réduit en groupe amino avec LiAlH4.
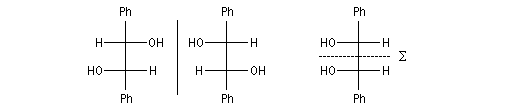
La réduction d'un composé dicarbonylé fournit au maximum quatre stéréo-isomères. Dans le cas du benzile, du fait de la symétrie de la molécule de départ, il y a deux énantiomères et un composé méso.
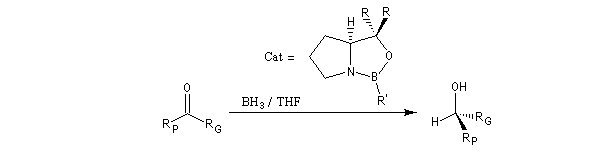
De nombreux réactifs permettant le transfert d'hydrure ont été préparés ces dernières années. Le K-sélectride est un hydrure fortement encombré utilisé dans les réductions
diastéréosélectives.
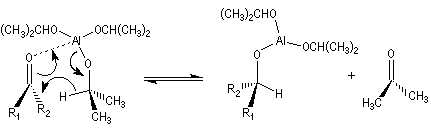
A ces réactions on peut rattacher la réaction de Meerwein, Ponndorf et Verley dans laquelle l'isopropylate d'aluminium permet le transfert d'un ion hydrure vers un composé carbonylé.
Les réactions de réduction des carbonylés impliquant des organomagnésiens encombrés procèdent d'un mécanisme assez comparable.
|
|
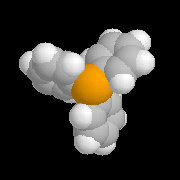
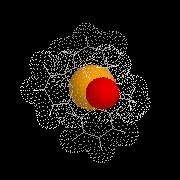
Le modèle ci-contre représente un hydrure du bore alkylé commercialisé sous le
nom de K-Sélectride (Aldrich). On l'obtient en faisant réagir un trialkylborane sur l'hydrure
de potassium.
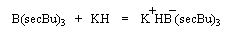
Il est beaucoup plus encombré et par conséquent plus stéréosélectif que le tétrahydroborate de
sodium lorsque la réaction est contrôlée par des facteurs stériques.
|
Hydratation
Résultats expérimentaux
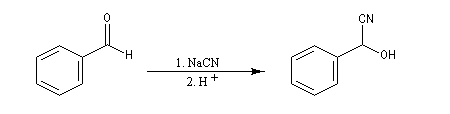
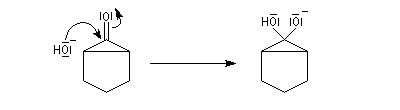
Le produit d'addition de l'eau sur un composé carbonylé est un gem-diol.
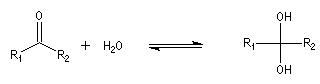
Si l'on excepte l'hydrate de chloral qu'on peut obtenir à l'état cristallisé, la plupart des gem-diols sont des composés instables.
D'un point de vue préparatif cette réaction offre donc peu d'intérêt. En revanche elle permet d'obtenir des informations intéressantes sur la réactivité comparée des composés carbonylés. Le tableau
ci-dessous regroupe quelques valeurs pour la constante thermodynamique K de l'équilibre.
K = [hydrate]/[carbonylé]
|
Composé |
CH3COCH3 |
CH3CHO |
HCHO |
F3CCHO |
|
K |
1,4´10-3 |
1,06 |
2,3´103 |
2,9´104 |
On constate que vis à vis de l'hydratation, les aldéhydes réagissent plus facilement et plus complètement que les
cétones. Cela peut s'expliquer par plusieurs facteurs :
- l'atome de carbone d'un aldéhyde est moins encombré que celui d'une cétone ;
- cet atome est moins substitué par des groupes donneurs par effet inductif. Il est plus déficient en électrons et donc plus électrophile ;
- notons la valeur élevée de la constante thermodynamique K pour le méthanal qui se distingue des autres aldéhydes par une plus grande réactivité.
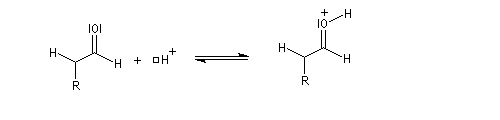
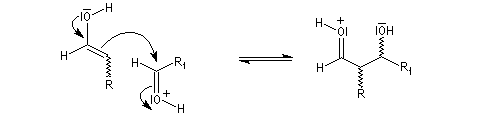
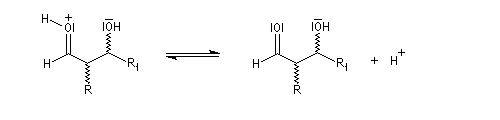
La réaction d'hydratation est catalysée en milieu acide et en milieu basique. Nous allons examiner successivement
les deux cas :
- catalyse acide ;
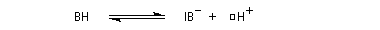
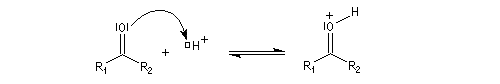
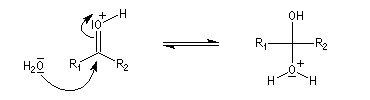
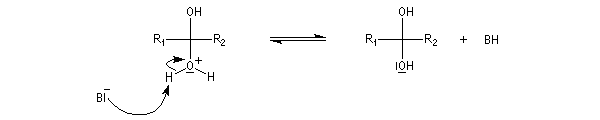
- catalyse basique.

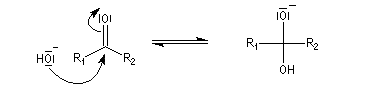
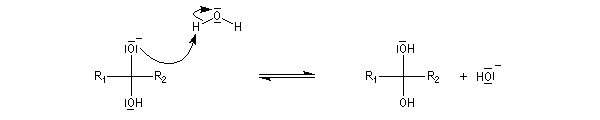
En réalité, la situation est un peu plus complexe car on constate que les acides et les bases, même
sous une forme non ionisée, catalysent cette réaction. On dit que la réaction est l'objet d'une catalyse acido-basique généralisée.
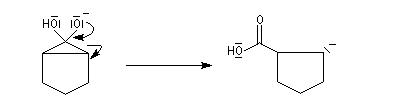
Oxydation des aldéhydes
On a pu démontrer que l'oxydation des aldéhydes en milieu aqueux fait intervenir l'hydrate du carbonylé. Un mécanisme
partiel de cette oxydation est le suivant :
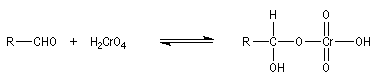
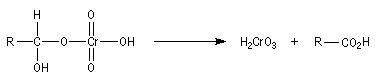
L'oxydation des alcools primaires en aldéhyde pose le problème de la suroxydation de ces
derniers en acide. Puisque l'oxydation implique l'hydrate de l'aldéhyde, un moyen de s'en
affranchir est de travailler en milieu anhydre. C'est le cas lorsqu'on effectue l'oxydation de
l'alcool en utilisant un réactif mis au point par le chimiste américain E. J. Corey : le chlorochromate de pyridinium PCC.
Addition des organométalliques
Généralités
La réaction entre un composé organométallique et un aldéhyde ou une cétone est généralement conduite en deux étapes :
- le composé carbonylé, dissous dans un solvant organique comme le toluène est mis à réagir avec l'organométallique ;
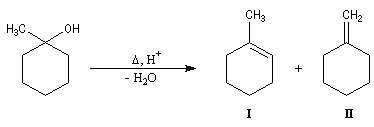
- le produit d'addition est un alcoolate magnésien. Cet alcoolate est hydrolysé en milieu acide afin d'éviter la précipitation de Mg(OH)2 . L'hydrolyse conduit à l'alcool. Dans la synthèse des alcools tertiaires il faut éviter d'utiliser un milieu trop acide afin d'éviter la déshydratation de l'alcool.
Une méthode découverte par Barbier, directeur de thèse de Grignard, consistait à mélanger le substrat, le réactif et le métal dans un même réacteur. Ce type de réaction monotope (single pot reaction) a connu récemment un regain d'intérêt. On réalise
de cette manière certaines réactions faisant intervenir le zinc sans que l'organozincique soit isolé. Un autre exemple de réaction au cours de laquelle l'organométallique n'est pas isolé est constitué par la réaction de Réformatsky.
Les a-énones donnent des réactions intéressantes avec différents organométalliques, notamment les organocuprates. Leurs propriétés sont étudiées dans un chapitre particulier.
Aldéhydes
La réaction entre un organomagnésien et un aldéhyde, suivie d'une hydrolyse acide, constitue une synthèse d'alcool secondaire. Voici quelques exemples.
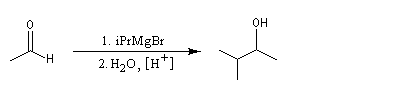
Notons, d'un point de vue expérimental, que les aldéhydes bouillent à des températures assez basses : 21 °C pour l'éthanal, - 21 °C pour le méthanal. Avec cet aldéhyde, l'alcool obtenu est primaire.
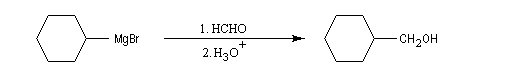
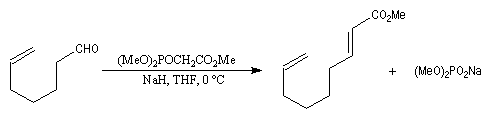
Les aldéhydes sont plus réactifs que les esters. La réaction suivante, effectuée à basse température, suivie d'une hydrolyse, est une étape de la synthèse du sulcatol, une phéromone d'insecte. La fonction ester sert ici à protéger une fonction alcool secondaire.
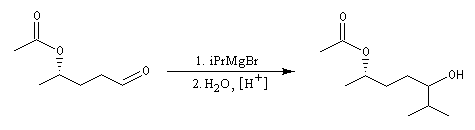
Cétones
Les cétones sont moins réactives que les aldéhydes à cause d'un carbone plus encombré et moins électrophile. La réaction entre un organomagnésien et une cétone conduit à un alcool tertiaire.
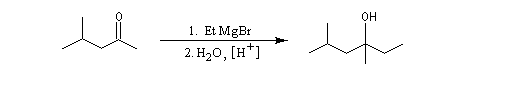
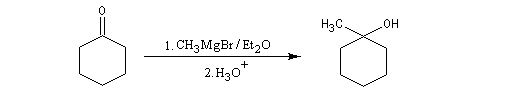
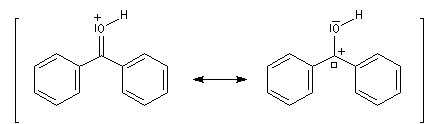
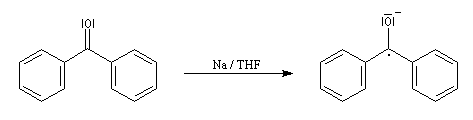
La réaction entre le bromure de phénylmagnésium et la benzophénone permet la préparation du triphénylméthanol.
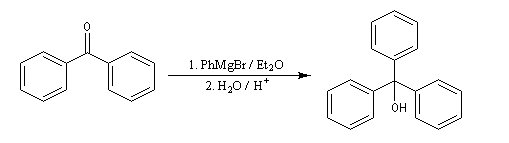
|

|
L'addition de benzophénone dans l'éther à une solution de bromure de phénylmagnésium, conduit dans un premier temps à une solution rouge traduisant la présence d'un intermédiaire réactionnel fortement délocalisé (un complexe de transfert de charge impliquant un radical anion).
L'hydrolyse acide de cette solution, dans une seconde étape, fournit principalement le triphénylméthanol (il peut également se former une petite quantité de benzopinacol par duplication des radicaux intermédiaires). Voir : [88]
|
Le tableau ci-dessous résume les différents cas possibles.
|
Composé |
méthanal |
aldéhydes |
cétones |
|
Classe d'alcool |
primaire |
secondaire |
tertiaire |
Mécanismes
Les mécanismes de réaction entre les organomagnésiens et les composés carbonylés ne sont pas complètement élucidés. Dans certains cas, la cinétique de la réaction est en faveur d'un mécanisme dans lequel intervient deux molécules d'organomagnésien (mais une loi cinétique ne permet pas de démontrer un mécanisme).
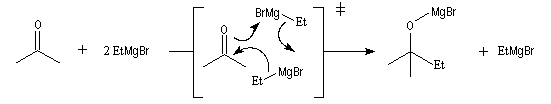
La réaction entre les organomagnésiens et les cétones aromatiques a été particulièrement étudiée par E. C. Ashby qui a pu démontrer l'intervention d'un mécanisme radicalaire.
Lorsqu'on ajoute lentement du bromure de phénylmagnésium à une solution de benzophénone dans le toluène, on observe de façon transitoire une coloration rouge. On l'interprète par l'existence d'un complexe de transfert de charge qui résulte d'un transfert monoélectronique du magnésien vers la cétone.
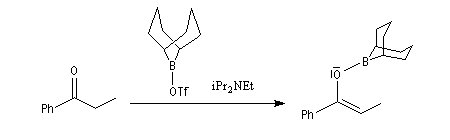
Aspect stéréochimique
La stéréochimie de l'addition d'un organométallique sur les faces diastéréotopiques
d'un composé carbonylé peut être prévue en utilisant le modèle de Felkin-Anh sauf quand la présence d'un groupe jouant le rôle de base de Lewis sur le substrat entraîne un contrôle par chélation de l'addition.
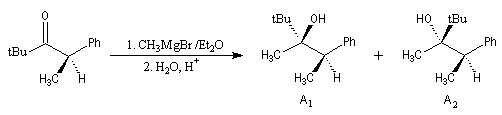
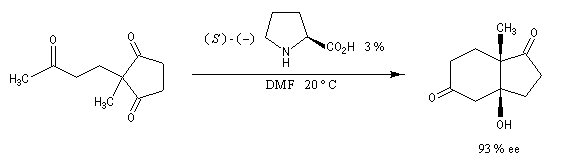
L'excès diastéréoisomérique en faveur du composé I, vaut 92 %.
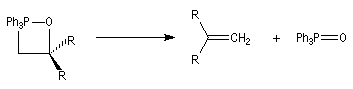
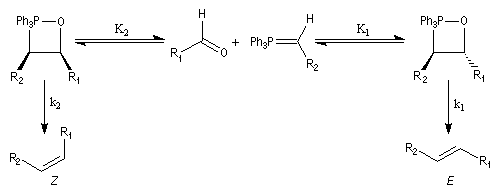
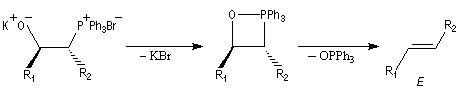
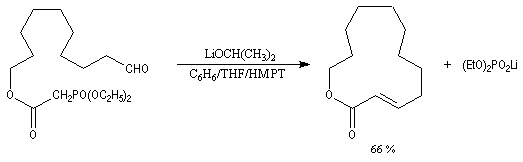
Réaction de Wittig
Introduction
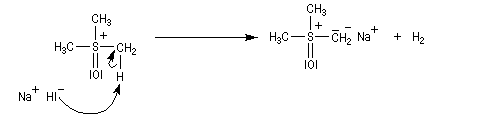
Le chimiste allemand G. Wittig qui est décédé en 1987, a travaillé dans de nombreux domaines de la chimie organique. Il est notamment connu pour la découverte de la réaction qui porte son nom et qui lui a valu le prix Nobel de chimie en 1979 [31] conjointement avec le chimiste américain H. C. Brown. En 1950, G. Wittig, qui était déjà l'auteur de nombreux travaux en chimie organométallique, cherchait à préparer des composés pentavalents de l'azote. L'équation de la réaction qu'il voulait réaliser s'écrit :
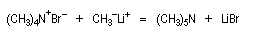
Inutile d'insister sur le fait qu'il n'obtint pas le produit convoité (!) mais un composé possédant une liaison carbone-azote provenant de la métallation d'une liaison C-H du substrat par le "At the time we were not sure whether hydrogen bound to carbon would
be proton-labile in quaternary ammonium salts. We came to this conclusion
with an absurd experiment to prepare pentamethylnitrogen from
tetramethylammonium salts by using the reaction of tetramethylammonium
halide with methyllithium.
It was confirmed experimentally that the octet principle is strictly valid for
the elements of the first eight-element period. The object of synthesizing
compounds with a pentacoordinate central atom was reached only when we
studied the higher elements of the fifth main group - that is, phosphorus,
arsenic, antimony, and bismuth." (extrait de la conférence donnée par G. Wittig à l'occasion de la remise de son prix Nobel en 1979) [68].
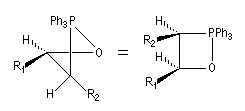
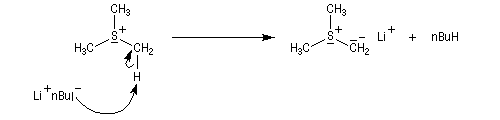
Tout naturellement Wittig se tourna ensuite vers l'élément phosphore dont l'aptitude à étendre son octet est bien connue dans des composés comme Notons que des calculs récents ont montré qu'une approche supra/supra est également possible si l'on tient compte de l'intervention des orbitales d du phosphore [79].