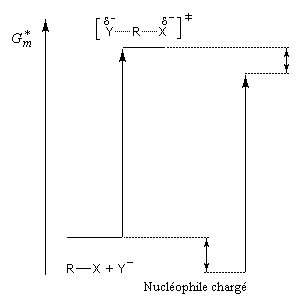
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Dérivés halogénés et substrats apparentés
Introduction
|
I |
II |
III |
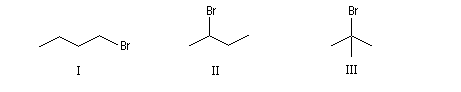
|
1-Bromobutane |
2-Bromobutane |
2-Méthyl-2-bromopropane |
Notons qu'il y a deux molécules de bromo-2-butane énantiomères.
Il existe des dérivés halogénés en série alicyclique ainsi que des composés dans lesquels l'halogène est relié à un groupe vinyle ou allyle.
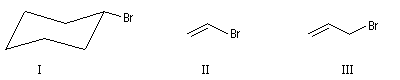
|
I |
II |
III |
|
Bromocyclohexane |
2-Bromoéthène |
3-bromopropène |
Certains aspects de la réactivité des dérivés halogénés aromatiques sont étudiés dans le chapitre relatif aux composés aromatiques. Ne sont signalées ici que les réactions de couplage catalysées par les métaux de transition comme le palladium.
Les dérivés halogénés sont des agents cancérogènes du fait de leur caractère alkylant. Les dérivés méthylés sont particulièrement toxiques et ne doivent être manipulés qu'avec beaucoup de précautions.Classe
Selon que l'atome de carbone portant l'halogène est lié à 1, 2, 3 atomes de carbone, le dérivé est qualifié de primaire, secondaire, tertiaire.
|
Dérivé halogéné |
Bromo-1-butane |
Bromo-2-butane |
2-Méthyl-2-bromopropane |
|
Classe |
I |
II |
III |
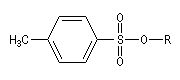
Les dérivés sulfonylés d'alcanes : mésylates, tosylates, triflates, remplaçent souvent les dérivés halogénés, en tant que substrats, dans de nombreuses réactions de substitutions nucléophiles. Ces composés sont quelquefois appelés pseudo-halogénés (pseudohalides en anglais).
|
|
Le pourpre qui ornait le bas de la toge romaine est un composé bromé extrait du murex, un coquillage abondant en Méditérannée. Ce colorant d'origine naturelle est connu depuis très longtemps puisque des textes en mentionnent l'existence en 1600 avant J.-C. La première synthèse de ce composé a été réalisée par Sachs et Kempf en 1903. On trouvera des informations intéressantes sur ce composé à la référence [20]. |
La thyroxine est une hormone présente dans la glande thyroïde.
Propriétés physiques
Caractéristiques physiques et énergétiques
|
Elément |
C |
F |
Cl |
Br |
I |
|
Electronégativité (Pauling) |
2,55 |
3,98 |
3,16 |
2,96 |
2,66 |
Températures de changement d’état
Le tableau suivant regroupe les températures de changement d'état des halogénures de méthyle.
On constate une augmentation du moment dipolaire lorsqu'on passe du dérivé iodé au dérivé fluoré. Cet accroissement est à mettre en relation avec l'augmentation de l'électronégativité de l'halogène. Cependant il faut faire attention que le moment dipolaire fait intervenir la longueur de liaison qui varie aussi.
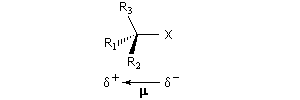
|
Composé |
CH3-H |
CH3-F |
CH3-Cl |
CH3-Br |
CH3-I |
|
TE (°C) |
-164 |
-78 |
-24 |
3 |
42 |
|
Longueur C-X (pm) |
101 |
135 |
177 |
194 |
214 |
|
Do (kJ.mol-1) |
435 |
485 |
327 |
285 |
213 |
|
Moment dipolaire m (D) |
0 |
1,92 |
2,05 |
2,01 |
1,87 |
L'élévation de la température d'ébullition quand on passe des dérivés fluorés aux dérivés iodés n'est pas seulement due à la variation du moment dipolaire mais aussi à l'augmentation de la polarisabilité des liaisons qui impliquent des atomes de plus en plus volumineux. Il y a donc augmentation des forces d'interaction de type :
Spectroscopie
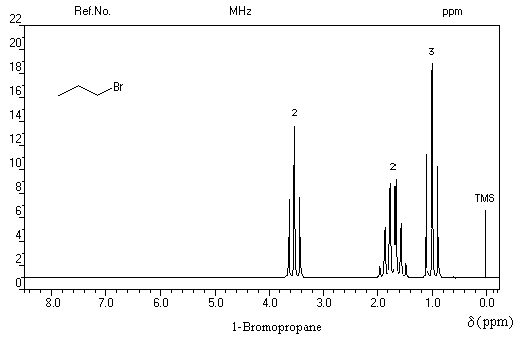
Spectroscopie infrarouge Spectroscopie de résonance magnétique nucléaire Composé CH3I CH3Br CH3Cl CH3F Déplacement chimique d (ppm) 2,3 2,7 3,4 4,5
Dans les composés suivants, on observe une augmentation du déplacement chimique avec l'accroissement de l'électronégativité de l'halogène. Ce phénomène est général. Plus l'halogène est l'électronégatif plus la densité électronique autour du noyau d'hydrogène est faible. L'intensité du champ nécessaire pour atteindre la résonance diminue. Il en résulte un accroissement du déplacement chimique.
Les spectres suivants illustrent la facilité avec laquelle on peut distinguer deux isomères de position par simple comparaison de leurs spectres de RMN grâce aux déplacements chimiques différents des groupes de protons et aux couplages entre-eux.
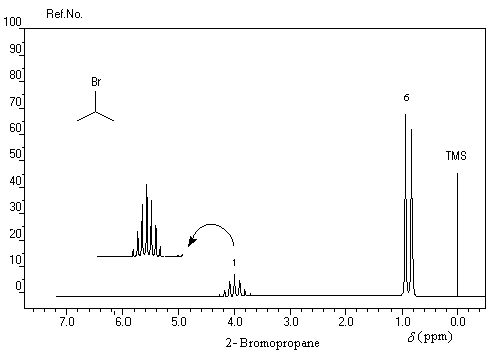
Substitutions nucléophiles
Définitions
Nous allons étudier des réactions de substitution c'est à dire des réactions au cours desquelles on observe la rupture d'une liaison et la reformation simultanée ou non d'une autre liaison.
Dans le cas qui nous occupe ici, le substrat est un halogénure d'alkyle. Le réactif est un nucléophile (étymologiquement : qui a une affinité pour les noyaux ; c'est à dire les zones déficientes en électrons). Ce terme se réfère à l'aptitude que possède une base de Lewis à céder son doublet. On peut résumer cela de façon formelle formelle par l'équation ci-dessous.
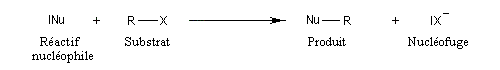
Remarque : les dérivés halogénés aromatiques ou vinyliques n'ont pas du tout la même réactivité que les halogénures d'alkyle dans les réactions de substitutions nucléophiles. Ils ne réagissent pas dans les conditions ordinaires. Ils donnent lieu à des réactions de couplage catalysées par des métaux de transition comme le palladium.
Substitutions nucléophiles bimoléculaires
Résultats expérimentaux, inversion de Walden
La réaction entre les ions iodure et (2S)-2-bromopropane, fournit le (2R)-2-iodopropane . La réaction s'accompagne d'une inversion de la configuration relative du centre chiral appelée inversion de Walden.
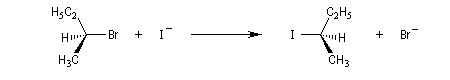
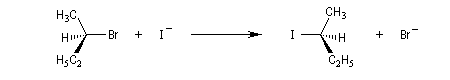
Dans les réactions précédentes le substrat et le produits n'étaient pas stéréoisomères. Mais lorsque le nucléophile et le nucléofuge sont les mêmes, le produit obtenu est l'énantiomère du substrat. L'évolution stéréochimique de la transformation suivante peut être faite grâce à l'utilisation d'ions iodure radioactifs.
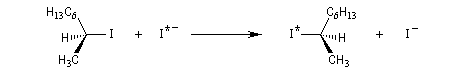
L'inversion de configuration d'un centre chiral au cours d'une substitution bimoléculaire porte le nom d'inversion de Walden du nom du chimiste allemand P. Walden qui l'a découverte en 1896 au cours d'une étude restée célèbre sur les énantiomères de l'acide malique.
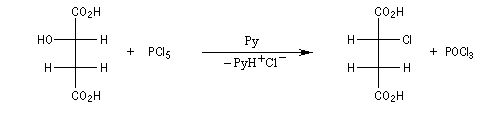
L'inversion de Walden peut naturellement avoir lieu sans modification de la configuration absolue. On trouvera un exemple de ce type dans le chapitre consacré aux règles de Cahn, Ingold, Prelog.
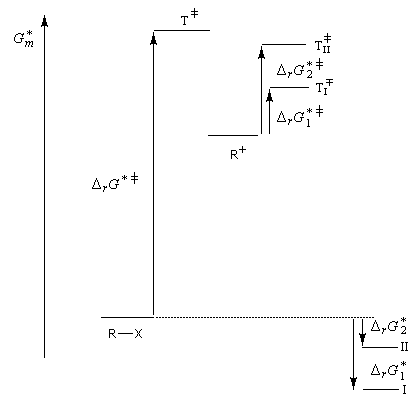
Mécanisme et cinétique
Le mécanisme admis est un choc bimoléculaire entre le nucléophile et le substrat. Il s'agit d'une réaction élémentaire.
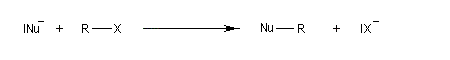
Par définition de la vitesse de la réaction.
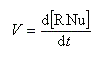
Pour une réaction élémentaire, l'ordre partiel par rapport à chacun des réactifs est égal au nombre stœchiométrique correspondant. La loi de vitesse s'écrit donc.
![]()
La molécularité est égale à deux. En l'absence de dégénérescence, cela se traduit expérimentalement par un ordre global égal à deux.
Puisque la transformation s'effectue en une seule étape élémentaire, le profil énergétique comporte un seul état de transition qui traduit la rupture partielle de la liaison entre l'atome de carbone et le nucléofuge et la formation partielle concomitante de la liaison entre l'atome de carbone et le nucléophile.
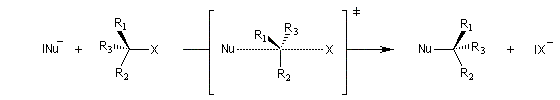
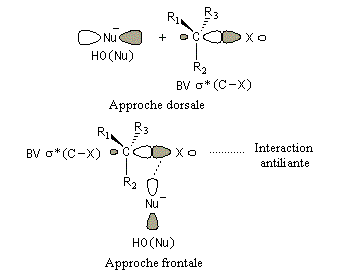
La réaction implique le recouvrement entre la plus haute orbitale occupée du nucléophile et la plus basse orbitale vacante du substrat.
|
|
Interprétation orbitalaire de l'inversion de Walden. Dans l'approximation des orbitales frontières, l'interaction principale a lieu entre la plus haute orbitale occupée du nucléophile (HO) et la plus basse orbitale vacante du substrat (BV) qui est l'orbitale antiliante de la liaison C-X.
|
En série cyclohexanique, l'attaque dorsale d'un dérivé substitué en position 4 du tertiobutylcyclohexane se traduit par la formation exclusive du diastéréoisomère de stéréochimie cis.
Nature du nucléophile
Cet aspect est étudié dans un chapitre particulier consacré aux bases de Lewis.
|
Liaison |
C-C |
C-F |
C-Cl |
C-Br |
C-I |
|
Vitesse relative |
- |
10-4 |
2,0´10-2 |
1,0 |
3,0 |
|
Do (kJ.mol-1) |
- |
485 |
340 |
285 |
213 |
|
Polarisabilité 1030´a (m-3) |
0,5 |
0,7 |
2,5 |
3,6 |
5,6 |
|
Nucléofugacité |
- |
1 |
2,0´102 |
104 |
3,0´104 |
La vitesse de la réaction augmente considérablement lorsqu'on passe du fluor à l'iode. Ce résultat est à mettre en relation avec l'accroissement de la polarisabilité de la liaison carbone-halogène. Une application intéressante de ces résultats est la réaction d'échange d'halogène (réaction de Finkelstein).
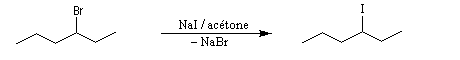
Effets stériques
Commençons par examiner l'influence de l'encombrement de l'atome de carbone en a de l'halogène (solvant : propanone à 298 K).
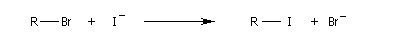
|
Composé |
CH3Br |
CH3CH2Br |
(CH3)2CHBr |
(CH3)3CBr |
|
Vitesse relative |
145 |
1,0 |
7,7´10-3 |
4,0´10-6 |
|
|
|
|
|
CH3CH2Br |
(CH3)2CHBr |
(CH3)3CBr |
Si l'encombrement de l'atome de carbone en b est important l'effet est encore très important. Les substrats néopentyliques réagissent très lentement.
|
Composé |
CH3CH2Br |
CH3(CH2)2Br |
(CH3)2CHCH2Br |
(CH3)3CCH2Br |
|
Vitesse relative |
1 |
0,8 |
3,0´10-3 |
1,3´10-5 |
Les effets stériques s'amortissent assez vite avec l'éloignement de l'atome de carbone fonctionnel.
|
Dérivé halogéné |
tBuCH2Br |
tBu(CH2)2Br |
tBu(CH2)3Br |
|
Vitesse relative |
1,3´10-5 |
3´10-2 |
1 |
|
|
La figure de gauche représente le 1-bromobicyclo[2,2,2]octane. Ce composé n'est pas du tout réactif dans les réactions de substitutions nucléophiles SN2 à cause de l'encombrement stérique de l'atome de carbone porteur de l'halogène. Il ne réagit pas non plus selon un processus SN1 car cela impliquerait la formation d'un carbocation non plan. |
Effets électroniques
On s'intéresse à la réaction suivante pour différents groupes R (solvant : acétone).
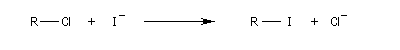
|
Composé |
CH3(CH2)2Cl |
CH2=CHCH2Cl |
PhCH2Cl |
CH3COCH2Cl |
|
Vitesse relative |
1,0 |
75 |
180 |
33 000 |
Les dérivés allyliques et benzyliques réagissent plus rapidement que les substrats saturés. On l'interprète par le fait que la charge qui se développe dans l'état de transition est stabilisée par résonance avec le système d'électrons p.
Notons qu'à du mécanisme SN2 ordinaire, il existe un mécanisme particulier nommé SN2' observé chez les systèmes allyliques.
Les composés carbonylés a-halogénés réagissent rapidement. La mobilité de l'halogène est accrue par l'interaction entre le nucléophile et le groupe carbonyle. Leur hydrolyse rapide dans les yeux provoque un comportement lacrymogène.
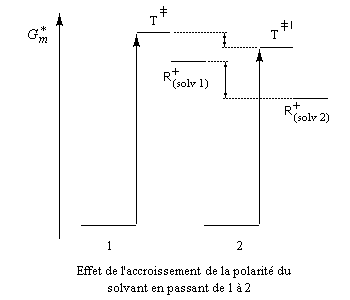
Influence du solvant
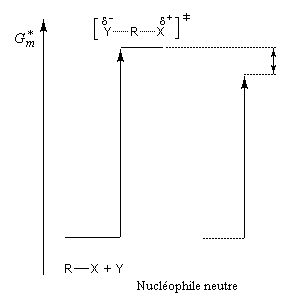
L'influence qualitative de la nature du solvant sur les substitutions nucléophiles a été étudiée par Hughes et Ingold. Le modèle consiste à comparer la solvatation des réactifs avec celle de état de transition. Nous nous limitons ici aux réactions SN2 faisant intervenir un dérivé halogéné comme substrat. Celui-ci est donc neutre.
La théorie de Hughes et Ingold fait intervenir seulement des interactions de nature électrostatique. Plus la solvatation d'une espèce par le solvant est grande, plus l'énergie de la nouvelle entité solvatée est abaissée. D'autre part, une espèce est d'autant plus solvatée que sa charge électrique est plus élevée et concentrée dans un volume plus petit.
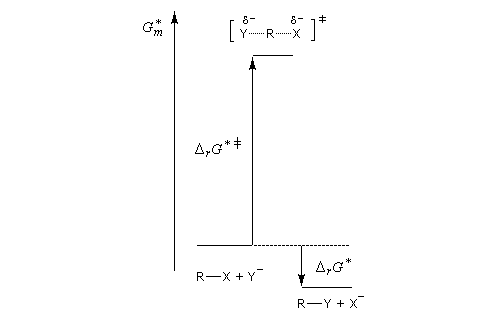
Puisque l'énergie d'activation est la différence entre l'énergie de l'état de transition et celle des réactifs, on en déduit :
Les diagrammes suivants montrent l'influence de l'augmentation de la polarité du solvant sur la vitesse de réaction.
|
|
|
|
Création de charges contraires dans l'état de transition. Augmentation de la vitesse |
Charge plus dispersée dans l'état de transition que dans les réactifs. Diminution de la vitesse |
Dans les solvants dipolaires aprotiques, on observe une grande accélération des réactions SN2 dans lesquelles le nucléophile est un anion. A titre d'exemple, la constante de vitesse de la réaction suivante est un million de fois plus grande dans le DMF que dans le méthanol.
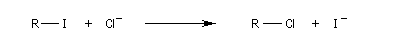
Les solvants dipolaires aprotiques se caractérisent par des dipôles dont les extrémités négatives sont tournées vers l'extérieur de la molécule de solvant. Au contraire, la partie positive de l'atome central est camouflée par les atomes périphériques. Il en résulte que ces solvants solvatent spécifiquement les cations. L'anion est libéré de la paire d'ions et il est peu ou pas solvaté. De ce fait, sa nucléophilie est considérablement exaltée par rapport à ce qu'on observe dans un solvant protique. Dans ce type de solvant, la nucléophilie des ions halogénures varie dans le même ordre que leur basicité :
I- > Br- > Cl-
|
Solvant |
Permittivité relative er |
Moment dipolaire |
|
38 |
3,8 |
|
|
49 |
4,3 |
|
|
30 |
5,5 |
L'utilisation d'agents de transfert de phase comme les ions ammonium quaternaires, les éthers couronnes et les cryptands permet d'exalter la nucléophilie des anions. On peut ainsi, par exemple, synthétiser des fluorures d'alkyles grâce à l'activation anionique obtenue par l'utilisation d'un éther couronne.
Applications diverses On trouvera ci-dessous une liste non exhaustive de réactions de substitution nucléophiles impliquant des réactifs organiques et inorganiques qui ont été traitées dans d'autres chapitres. Dans la réaction suivante on note l'inversion de Walden du centre chiral porteur du nucléofuge. Les substitutions nucléophiles peuvent avoir lieu de façon intramoléculaire. On trouve des exemples dans la synthèse de Williamson des éthers et en particulier des époxydes. La réaction de Darzens dans sa dernière étape constitue un cas particulier de substitution intramoléculaire. Des substitutions nucléophiles intramoléculaires peuvent intervenir avec rétention de configuration. Un exemple de tel mécanisme SNi s'observe lors de la réaction entre un alcool et le chlorure de thionyle dans l'éther en l'absence de pyridine. Dérivés sulfonylés Les nucléofuges sont des bases beaucoup plus faibles donc plus facilement déplaçables que les ions halogénures. Les triflates d'alkyle sont, à cet égard, d'excellents substrats car l'ion triflate est la base conjuguée, très stable, de l'acide triflique, un superacide. Mésylate (ROMs) Triflate (ROTf) Tosylate (ROTs)
Nous allons décrire dans ce paragraphe quelques exemples d'alkylation de réactifs inorganiques. Le premier exemple est la synthèse des nitriles de Kolbe.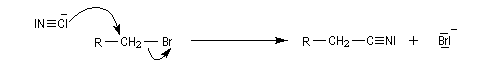
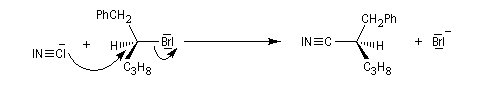
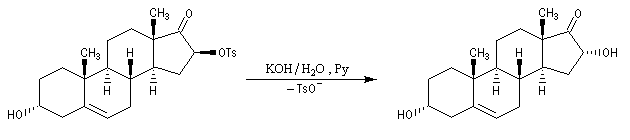
Notons que dans ce type de réaction, les dérivés halogénés sont avantageusement remplacés en tant que substrats par des mésylates, des tosylates ou des triflates obtenus par sulfonylation des alcools.
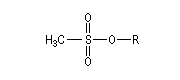
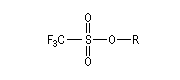
Substitutions nucléophiles monomoléculaires
Introduction
Pour un certain nombre de réactions de substitutions, on observe les résultats expérimentaux suivants :
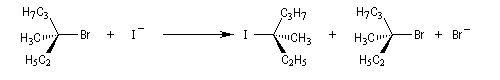
Mécanisme et cinétique
Le mécanisme suivant a été proposé :
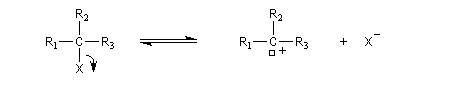
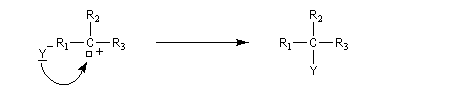
Introduisons les constantes de vitesse de chacune des étapes.
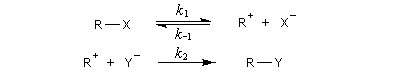
La première étape est cinétiquement déterminante. Elle est monomoléculaire et donne son nom au mécanisme SN1.
Par définition, la vitesse de la réaction est :
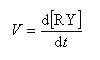
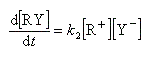
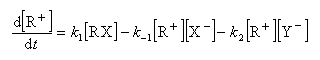
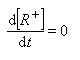
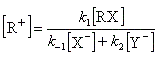
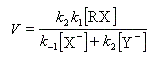
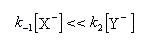
Le profil énergétique de la réaction est le suivant.
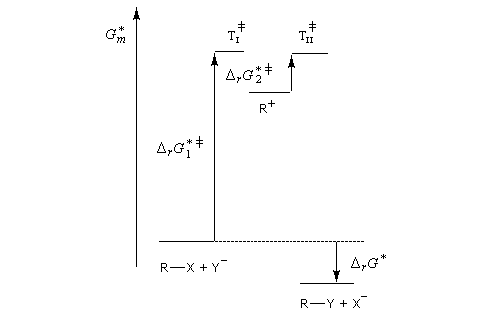
L'étape d'ionisation du substrat étant cinétiquement déterminante, c'est elle qui possède la plus grande énergie d'activation (état de transition tardif). En revanche, l'étape au cours de laquelle le nucléophile réagit avec le carbocation possède une énergie d'activation faible (état de transition précoce).
Puisqu'il se forme un intermédiaire de haute énergie, on peut appliquer le postulat de Hammond. Celui-ci nous indique que les deux états de transition ayant une énergie proche de celle du carbocation auront une structure voisine de celui-ci. En conséquence, tout facteur stabilisant le carbocation abaisse l'énergie de l'état de transition qui y conduit, donc accroit la vitesse de la réaction.
Effets stériques
Le passage de la géométrie tétraédrique de l'atome de carbone d'un dérivé halogéné à la géométrie plane de l'atome de carbone d'un carbocation s'accompagne d'une diminution des répulsions entre les liaisons car les angles entre-elles passsent de 109 ° à 120 °. Cette décompression stérique est maximale
lorsque le dérivé halogéné est tertiaire.
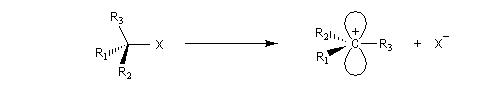
Dans certains systèmes pontés l'atome de carbone ne peut adopter une géométrie localement plane du fait de contraintes stériques importantes. L'exemple suivant illustre cette situation. Pour une raison analogue, on ne peut pas former facilement de double liaison en tête de pont (règle de Bredt).
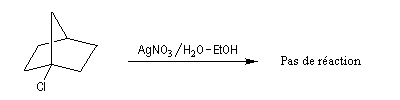
|
|
La figure de gauche représente le 1-bromobicyclo[2,2,1]heptane ou 1-bromonorbornane. Lorsqu'on mélange ce composé avec une solution hydroalcoolique de nitrate d'argent, on n'observe pas de réaction même à chaud. Cela montre que ce composé n'est pas du tout réactif dans les réactions de substitutions nucléophiles SN1 car cela conduirait à un carbocation non plan. Notons que ce composé ne réagit pas non plus selon un processus SN2 à cause de l'encombrement stérique de l'atome de carbone porteur de l'halogène. |
Effets électroniques
On s'intéresse aux vitesses relatives de solvolyse dans l'acide méthanoïque pour la réaction suivante (100 °C).
Les résultats obtenus avec différents substrats sont donnés ci-dessous.
|
Composé |
CH3Br |
CH3CH2Br |
(CH3)2CHBr |
(CH3)3CBr |
|
Vitesse relative |
0,6 |
1,0 |
25 |
108 |
On constate que la vitesse est d'autant plus grande que l'atome de carbone portant l'halogène est substitué par plus de groupes alkyles. Ce résultat s'interprète par le fait que l'énergie du carbocation formé est d'autant plus petite que celui-ci est davantage substitué par ces groupes. Ainsi qu'on l'a vu plus haut lors de l'étude du mécanisme, le postulat de Hammond permet de dire que les deux états de transition ayant une énergie proche de celle du carbocation auront une structure voisine de celui-ci. En conséquence, le carbocation le plus stable correspond à l'énergie de l'état de transition la plus basse. Le diagramme suivant résume cette situation.
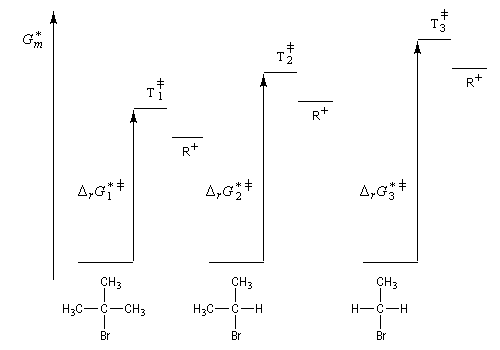
Schématiquement, l'augmentation du nombre de substituants alkyles augmente la stabilité des carbocations pour deux raisons :
Les halogénures vinyliques sont très peu réactifs vis à vis des réactions de substitution nucléophile. De même, les halogénures aromatiques ne réagissent pas dans les conditions ordinaires. Pour créer des liaisons CC avec ces substrats, on utilise des réactions de couplage catalysées par des métaux de transition comme le palladium.
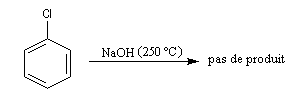
L'halogène étant donneur mésomère, on assiste à un renforcement de la liaison carbone-halogène. Les formes mésomères ci-dessous traduisent ce phénomène.
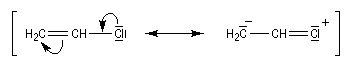
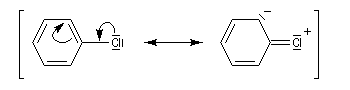
Au contraire, les substrats possèdant une liaison multiple sur l'atome de carbone en b, sont beaucoup plus réactifs que les substrats saturés.
|
Composé |
CH3CH2OTs |
iPrOTs |
CH2=CH-CH2OTs |
PhCH2OTs |
|
Vitesse relative |
1 |
2,7 |
33 |
385 |
Les systèmes allyliques réagissent généralement selon un mécanisme SN1 car ils peuvent conduirent à la formation de carbocations stabilisés.
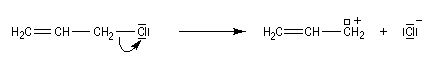
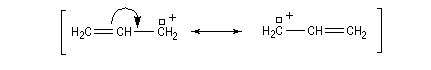
La réaction peut s'accompagner d'une réaction de transposition allylique. Notons également l'existence d'un mécanisme bimoléculaire plus rare, noté SN2'.
Les systèmes benzyliques se comportent de façon comparable. Les possibilités de délocalisation sont encore plus élevées.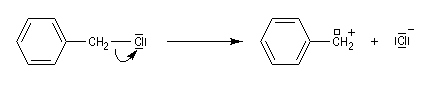
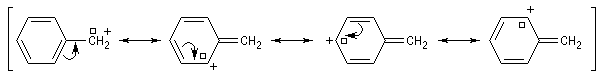
Influence du solvant
Afin d'étudier l'influence du solvant, il faut examiner comment il intervient dans l'étape d'ionisation du substrat qui est cinétiquement déterminante :
![]()
Le tableau suivant regroupe quelques résultats expérimentaux. On constate que l'acide méthanoïque (formique) et l'eau sont de très bons solvants pour ce type de réaction.
|
Solvant |
Permittivité relative er |
Constante de vitesse relative k |
|
CH3CO2H |
6 |
1 |
|
CH3OH |
33 |
4,0 |
|
HCO2H |
58 |
5,0´103 |
|
H2O |
78 |
1,5´105 |
Action des acides de Lewis
L'ajout d'acides de Lewis comme Ag+ ou AlCl3 qui forment des complexes acide-base de Lewis avec l'ion halogénure facilite l'ionisation du substrat. La vitesse de la réaction avec un nucléophile est accrue.
![]()
![]()
Si l'on ajoute une solution hydro-alcoolique de nitrate d'argent à un dérivé halogéné, on observe la formation d'un trouble qui traduit l'apparition de nouvelles espèces. Cela constituait naguère un test simple (mais peu fiable) de la classe des dérivés halogénés.
|
Classe du dérivé halogéné |
I |
II |
III |
|
Réaction |
lente à chaud |
lente à la température ambiante |
instantanée à la température ambiante |
Le développement d'une charge positive sur l'atome de carbone du substrat en présence d'un acide de Lewis augmente sa dureté. Cet effet est visible lorsqu'on utilise un nucléophile ambident.
La réaction "normale" entre un dérivé halogéné et un ion cyanure conduit à un nitrile par un mécanisme de type SN2. L'ion cyanure réagit par son pôle carboné mou, sur l'atome de carbone mou du substrat.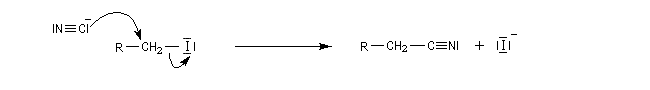
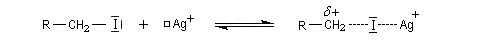
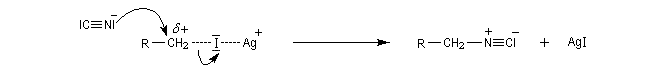
Stéréochimie
Dans l'introduction à l'étude des substitutions nucléophiles SN1, nous avons vu qu'en partant d'un substrat chiral, ces réactions conduisent généralement à un mélange racémique.
Un mécanisme plus fidèle à la réalité fait intervenir des paires d'ions. Dans ce qui suit, (s) désigne le solvant.
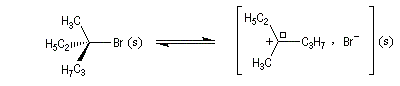
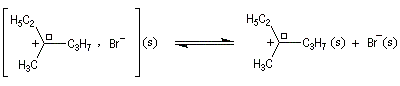
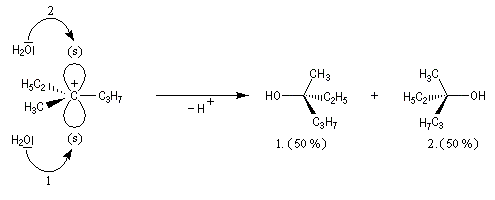
Si l'attaque du nucléophile a lieu sur le carbocation symétriquement solvaté, elle est équiprobable sur chacune des faces.
Selon les conditions expérimentales, et la nature du nucléofuge, le nucléophile peut réagir sur un cation non symétriquement solvaté. Dans ce cas, l'attaque sur la face opposée au nucléofuge qui est la plus dégagée est plus probable. On observe donc un excès du produit dont la configuration est inversée par rapport au substrat de départ.
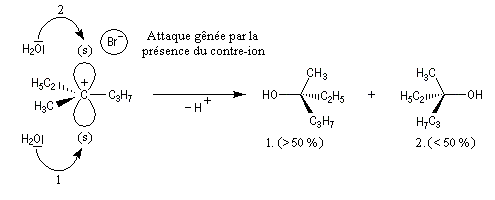
Examinons à présent le cas où le substrat possède deux centres chiraux dont un seul est susceptible d'être ionisé.
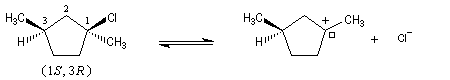
Le carbocation présente de faces diastéréotopiques non équivalentes. On obtient des produits diastéréo-isomères en proportions inégales. Le centre chiral non affecté par la substitution exerce une induction asymétrique.
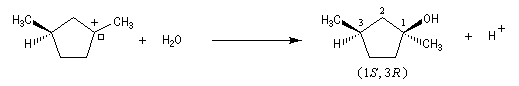
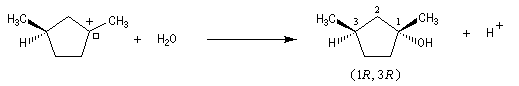
Dans certains cas, l'une des faces de la molécule est beaucoup plus encombrée que l'autre. Les proportions de produits obtenus peuvent alors être très différentes. Dans l'exemple suivant on obtient le produit chiral avec un rendement de 85 %.
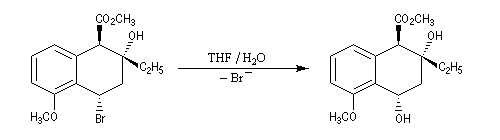
Des cas plus complexes ont été mis en évidence lorsque intervient la participation d'un groupement voisin.
Transpositions de carbocations
Dans une réaction de transposition, un atome ou un groupe d'atomes se déplace d'un point à un autre. Lorsqu'il y a migration d'une liaison sigma, il s'agit d'une transposition sigmatropique.
![]()
La force motrice de la transposition est la formation d'un carbocation plus stable.
![]()
![]()
Ce type de réaction est un cas particulier de transposition de Wagner-Meerwein.
Puisque les carbocations peuvent être générés facilement à partir d'alcools tertiaires, on rencontre là aussi ce type de transposition.
.Dans une réaction d'élimination, deux groupes appartenant à une molécule et généralement portés par des atomes différents, sont éliminés (les a-éliminations constituent un cas particulier). Il y a formation d'une insaturation. Celle-ci peut être une liaison double ou correspondre à un cycle.
Eliminations b bimoléculaires E2
Introduction
Les réactions d'élimination b initiées par des bases à partir des dérivés halogénés produisent des alcènes. On peut les regarder comme les réactions inverses des additions électrophiles des halogénures d'hydrogène sur la double liaison éthylénique.
Stéréosélectivité, stéréospécificité
La réaction d'élimination nucléophile bimoléculaire est diastéréospécifique. Afin que les orbitales à l'origine de la double liaison puissent se recouvrir efficacement, les liaisons impliquées dans le processus d'élimination doivent être dans un même plan. Lorsque les contraintes au sein de la molécule
le permettent, la disposition favorable des liaisons correspond à une géométrie antipériplanaire.
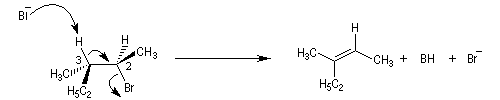
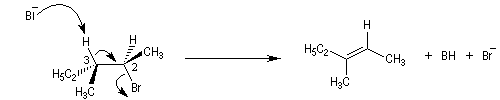
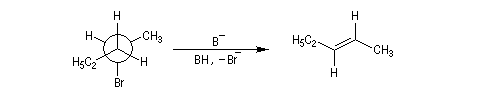
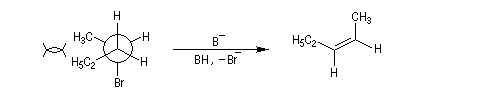
On obtient donc de façon majoritaire le diastéréo-isomère de configuration E.
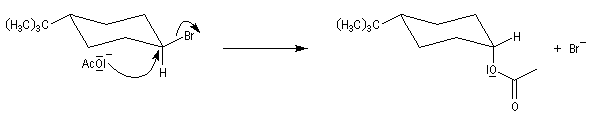
En série cyclohexanique, le fait que les liaisons doivent adopter une position antipériplanaire implique que les liaisons soient diaxiales. Une molécule rigide comme un dérivé bromé du tertiobutylcyclohexane en a du groupe tertiobutyle, ne conduit à une élimination facile qu'avec le dérivé cis.
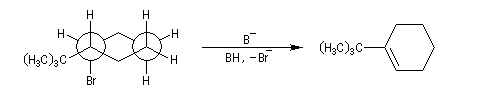
Lorsque l'équilibre entre plusieurs conformations est possible, l'élimination s'effectue sur une conformation trans diaxiale bien que la conformation diéquatoriale soit plus stable. Le basculement conformationnel d'une conformation à l'autre ne nécessite qu'une faible énergie. Ce résultat est conforme au principe de Curtin-Hammett.
Un autre exemple concerne l'élimination sur le chlorure de menthyle (I) et le chlorure de néomenthyle (II).
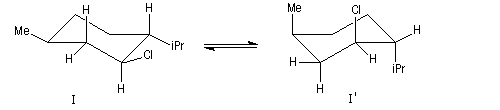
Dans le cas du chlorure de menthyle, l'élimination est difficile car elle s'effectue sur la conformation I' peu stable du fait que les substituants sont en position axiale.
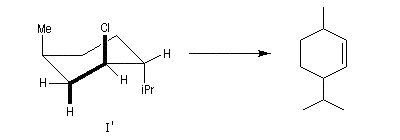
Dans le chlorure de néomenthyle (II) l'élimination est beaucoup plus facile car un seul substituant est en position axiale. Elle conduit de façon majoritaire au composé
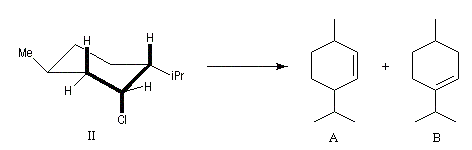
Dans certains composés composés pontés comme 1-chlorobicyclo[2,2,1]heptane exo (chlorure de bornyle), la rigidité du système bicyclique s'oppose à une disposition antipériplanaire des liaisons. On observe alors une élimination syn.
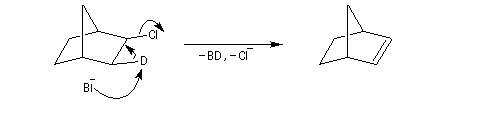
On connait d'autres exemples d'élimination syn. Citons-en quelques-uns :
Régiosélectivité, règle de Zaytzev
La transformation étant irréversible, les déshydrohalogénations E2 sont sous contrôle cinétique. Le produit majoritaire est le plus vite formé. On observe que le composé éthylénique le plus substitué est majoritaire car la double liaison est déjà partiellement formée dans l'état de transition (règle empirique de Zaytzev).
Ces résultats peuvent être rationnalisés par la théorie de l'état de transition variable.
Influence de la nature de la base
Pour promouvoir les éliminations E2 on utilise des bases fortes (HO-, RO-, H2N- ). On a aussi recours à des amines encombrées non nucléophiles (DBU, DBN, DABCO).
Influence du solvant
Il ne faut pas utiliser de solvant trop protique de façon à laisser à la base sa pleine capacité d'action. C'est la raison pour laquelle on utilise de préférence KOH dissous dans l'éthanol plutôt que dans l'eau. Certaines composés ioniques comme tBuOK voient leur activité basique renforcée par l'utilisation d'agents de transfert de phase comme les éthers couronnes ou les cryptands. La complexation de l'ion K+ exalte la basicité de l'anion.
Compétition entre les mécanismes E2 et SN2
L'élimination E2 et la substitution SN2 sont toujours en compétition l'une avec l'autre. Examinons quelques résultats expérimentaux.
|
Dérivé halogéné |
Pourcentage de substitution |
Pourcentage d'élimination |
|
CH3CH2Br |
100 |
0 |
|
(CH3)2CHBr |
100 |
0 |
|
(CH3)3CBr |
0 |
100 |
|
(CH3)2CHCHBrCH3 |
11 |
89 |
|
Dérivé halogéné |
Pourcentage de substitution |
Pourcentage d'élimination |
|
CH3CH2Br |
99 |
1 |
|
(CH3)2CHBr |
20 |
80 |
|
(CH3)2CHCH2Br |
40 |
60 |
|
CH3CH2 CHBrCH2CH3 |
12 |
88 |
Il ressort de ces valeurs numériques les résultats suivants :
Applications
Lorsqu'on réalise une réaction d'élimination sur un dérivé dihalogéné, on obtient dans un premier temps un halogénure vinylique. Si l'on poursuit la réaction, on peut obtenir un diène 1,3. L'exemple suivant est une étape de la synthèse de Schenk de la cantharidine.
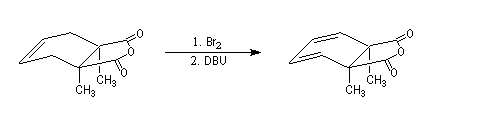
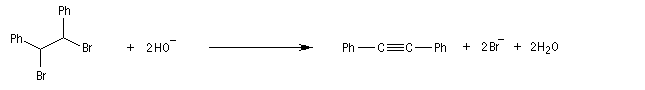
Il se forme dans un premier temps un halogénure vinylique.
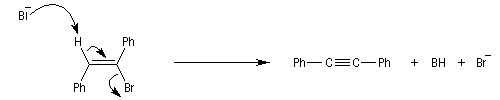
De même, l'élimination a partir d'un dérivé gem-dihalogéné, fournit un alcyne.
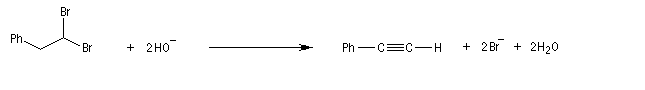
Eliminations b monomoléculaires E1
Introduction
On effectue la solvolyse du 2-bromo-2-méthylpropane (bromure de tertiobutyle) dans un mélange éthanol-eau. A côté du produit de substitution normalement attendu qui est l'alcool, on observe
la formation d'un composé éthylénique. Ce dernier produit est le fruit d'une réaction d'élimination.
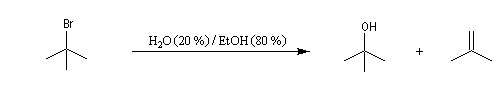
![]()
Mécanisme
Le mécanisme suivant en deux stades permet de rendre compte des résultats expérimentaux :
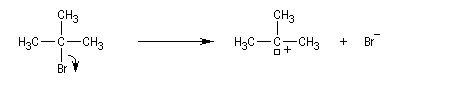
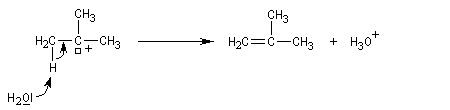
Ce mécanisme fait intervenir une première étape cinétiquement déterminante identique à celle qu'on observe dans le mécanisme SN1.
Régiosélectivité
On s'intéresse à la solvolyse du 2-bromo-2-méthylbutane dans un mélange éthanol eau 80/20.
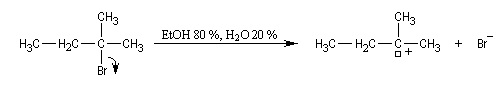
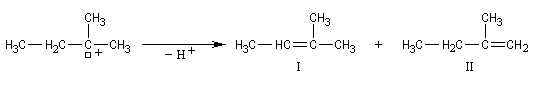
A cela, il faut ajouter les composés issus d'une substitution SN1.
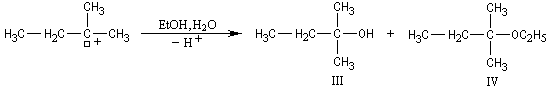
On observe les pourcentages suivants.
|
Dérivé halogéné |
I |
II |
III + IV |
|
Pourcentage |
32 |
8 |
60 |
Ces résultats montrent que le rapport des pourcentages en composés éthyléniques I/II = 4, n'est pas celui qui résulterait d'un équilibre entre ceux-ci. On est ici dans une situation différente de celle qu'on observe dans la déshydratation des alcools pour laquelle on observe l'équilibre suivant.
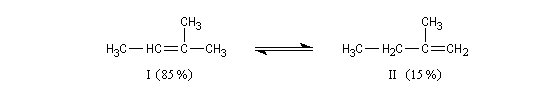
Cela signifie que la régiosélectivité observée est le résultat d'un contrôle cinétique et non thermodynamique de la transformation. Le composé éthylénique majoritaire est celui qui est formé le plus vite. L'état de transition qui y conduit est celui qui possède l'enthalpie libre molaire d'activation la plus basse. On interprète ce résultat par le fait que la formation de la liaison double est déjà commencée dans l'état de transition.
Notons que l'alcène le moins substitué peut être majoritaire lorsque des contraintes stériques importantes s'opposent à la formation de l'alcène le plus substitué.
Compétition entre les mécanismes E1 et SN1
Puisque la première étape de leur mécanisme respectif est commune, l'élimination E1 est toujours en compétition avec la substitution SN1. Les facteurs qui influencent l'un ou l'autre des deux mécanismes sont assez semblables à ceux qui interviennent
pour déterminer le rapport SN2/E2. L'élimination est favorisée par :
Dans les applications, il est très rare qu'on cherche à promouvoir des éliminations de type E1 chez les dérivés halogénés. La plupart du temps cette élimination est une réaction secondaire observée à côté de la substitution.
Eliminations b E1cB
Introduction
Lorsqu'on fait réagir le 1,1-dichloro-2,2,2-trifluoroéthane avec une solution aqueuse d'hydroxyde de sodium, on observe la réaction suivante.
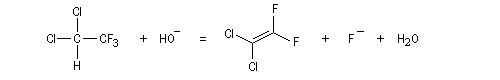
La cinétique est d'ordre global deux. Ordre partiel un par rapport à l'ion hydroxyde et par rapport à l'halogénure. Lorsque la réaction est effectuée dans l'eau lourde, on retrouve une partie du substrat deutérié. Cela indique qu'il s'est formé un carbanion intermédiaire. Dans l'exemple cité, l'arrachage du proton par la base pour former le carbanion est réversible.
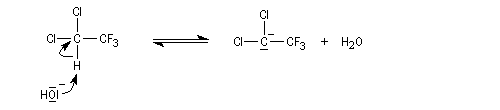
L'élimination de l'ion halogénure à partir de la base conjuguée (en anglais : conjugate base d'où cB) du substrat intervient dans la seconde étape. Cette étape monomoléculaire est cinétiquement déterminante.
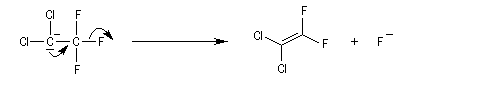
Le mécanisme précédent est appelé (E1cB)R (R comme réversible). Dans d'autres cas, l'étape cinétiquement déterminante est l'arrachage du proton par la base. On a alors un mécanisme (E1cB)I (I comme irréversible). Quoi qu'il en soit, le mécanisme E1cB nécessite la formation d'un carbanion suffisamment stable et un mauvais nucléofuge. On le rencontre dans la crotonisation des aldols en milieu basique et dans la condensation de Knoevenagel.
Théorie de l'état de transition variable
Les résultats expérimentaux montrent que l'élimination E2 fait intervenir un ensemble de mécanismes qui diffèrent les uns des autres par la synchronisation plus ou moins élevée des ruptures des liaisons
C-H et C-X. Dans la théorie de l'état de transition variable développée par le chimiste américain J. F. Bunnett, le mécanisme E2 synchrone occupe le centre du spectre tandis que les limites correspondent aux mécanismes E1Cb et E1 respectivement [13].
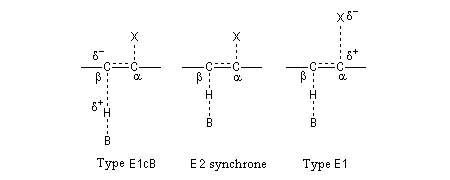
La position de la réaction dans le spectre fait intervenir plusieurs facteurs :
Autres réactions d'élimination
a-élimination
Dans une réaction d'a-élimination, les deux groupes éliminés sont liés au même atome de carbone. Il y a formation d'un carbène (ou d'un intermédiaire analogue).
L'existence des carbènes comme intermédiaires de réaction a été postulée par J. Hine en 1950 lors de son étude de la réaction entre le chloroforme et les ions hydroxyde [15].
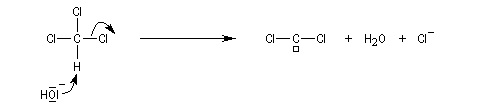
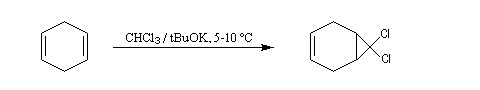
Une autre méthode ce cyclopropanation consiste à utiliser le couple CH2I2, Zn-Cu (réaction de Simmons-Smith).
La réaction de Reimer-Tiemann constitue un autre exemple d'utilisation de dihalogénocarbènes en synthèse.
Notons que des dérivés organométalliques de carbènes interviennent comme intermédiaires dans la métathèse des composés éthyléniques.
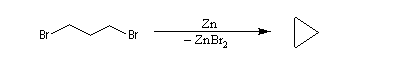
Dans la réaction suivante, la fonction nitrile permet la formation d'un carbanion stabilisé. On peut regarder la regarder comme une alkylation de ce carbanion par substitution nucléophile interne. Cette réaction constitue une préparation d'un précurseur de l'acide cyclopropanecarboxylique [29].
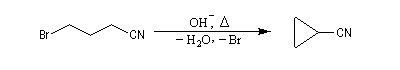
d-élimination
Des électrons de liaisons multiples peuvent assurer un relais dans le déplacement des liaisons. La réaction suivante est une méthode de synthèse de diènes conjugués. Une autre voie d'accès à ces composés consiste à effectuer deux éliminations b successives.
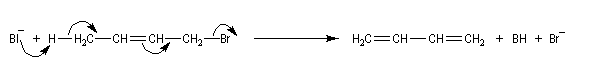
Elimination-décarboxylation
Il s'agit d'une réaction d'élimination des ions carboxylate des acides b-halogénés. Elle est utilisée dans la synthèse stéréospécifique des composés éthyléniques.
Elimination utilisant des métaux
Les dérivés dihalogénés traités par un métal comme le zinc ou le magnésium conduisent au composé éthylénique par une réaction d'élimination. Il y a formation d'un composé organométallique intermédiaire.
L'exemple suivant concerne la synthèse du cyclobutène [34].
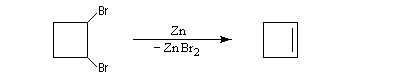
Une réaction du même type permet de synthétiser le plus simple des allènes [33].
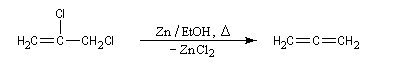
On peut mettre à profit cette réaction pour régénérer une double liaison éthylénique après protection par addition de dibrome. La réaction entre le magnésium et le dibromo-1,2-éthane peut servir à activer le magnésium dans la synthèse des organomagnésiens.
A partir du 1,3-dibromopropane, on obtient le cyclopropane par une réaction de g-élimination.
Couplages utilisant des métaux de transition comme catalyseurs
Introduction
L'importance des réactions de couplage catalytique des dérivés halogénés utilisant des complexes de métaux de transition comme ceux du palladium, a été récemment illustrée par l'attribution du prix Nobel de chimie 2010 au chimiste américain R. Heck et aux chimistes japonais Negishi et Suzuki. Historiquement, la première réaction de couplage croisé à avoir été publiée en 1972 est la réaction de Kumada entre un dérivé halogéné et un organomagnésien.
Réaction de Mozoroki-Heck
Cette réaction a été découverte indépendamment par le chimiste japonais T. Mizoroki et l'américain R. Heck dans les années 1970 [16].
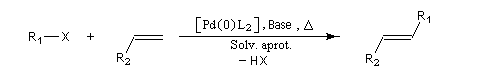
Le composé éthylénique doit posséder au moins un atome d'hydrogène vinylique.
Le catalyseur est un complexe du palladium (0) comme le tris(dibenzylidèneacétone)dipalladium (0) Pd2(dba)3 qui est une source stable de Pd (0). On peut aussi utiliser un composé du Pd (II) tel que Pd(OAc)2 en présence d'une phosphine telle que PPh3 permettant de générer Pd (0) in situ.
Un solvant aprotique tel que (THF, DMF) (mais des procédures en milieu aqueux existent)
La base est typiquement une amine tertiaire telle que Et3N.
La température est comprise dans la gamme : 20 °C à 140 °C.
La réaction est stéréosélective. On obtient de façon préférentielle le composé éthylénique de stéréochimie E. L'aspect mécanistique de la réaction de Heck est différent de celui des réactions de Suzuki et de Negishi. Il existe en fait deux mécanismes possibles :
La réaction a été utilisée dans la synthèse de nombreux composés cycliques complexes comme la morphine ou le taxol [40]. Elle est utilisée dans l'industrie pharmaceutique pour la synthèse du Naproxène.
Réaction de Negishi
Cette réaction été mise au point par le chimiste japonais Negishi en 1977 [50]. Celui-ci a obtenu le prix Nobel de chimie en 2010 pour cette découverte [36].
C'est une réaction de couplage croisé catalysée par un complexe du palladium, entre un dérivé halogéné R1X et un organozincique R2ZnX'.
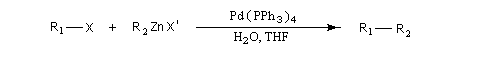
Les conditions de la réaction sont très douces. Elle est effectuée à la température ambiante.
La présence du zinc permet à la réaction d'être compatible avec de nombreux groupes fonctionnels (alcool , phénol, silanes, acétates, ester boroniques, amides).
Le mécanisme simplifié est résumé ci-dessous.
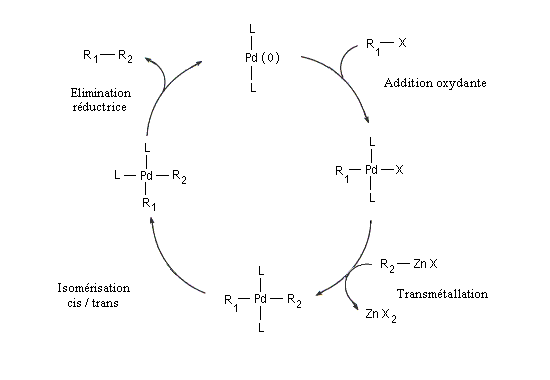
Il comporte les étapes suivantes :
La réaction de Negishi a notamment été utilisée avec succès pour synthétiser des biaryles non symétriques encombrés qui sont traditionnellement difficiles d'accès.La réaction suivante est une variante énantiosélective utilisant comme catalyseur, le tris(dibenzylidèneacétone)dipalladium (0) et comme catalyseur chiral un dérivé du ferrocène. Elle permet la préparation d'un binaphtyle chiral avec une très bonne énantiosélectivité. (P. Espinet et coll.)
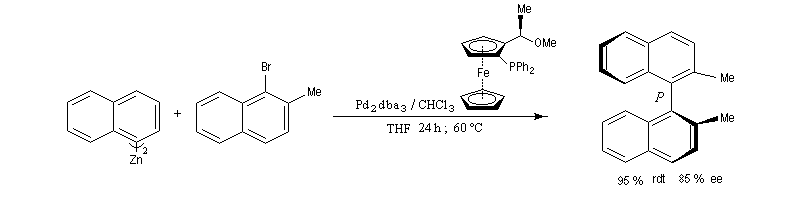
Notons cependant que l'un des inconvénients habituels de la réaction de Negishi est qu'elle nécessite la préparation in situ de l'organozincique. Ce dernier non isolable n'est donc pas facile à purifier.
Réaction de Suzuki
Cette réaction a été proposée par N. Miyaura et A. Suzuki en 1979 [51]. Ce dernier a obtenu le prix Nobel de chimie en 2010 pour cette découverte [36].
Il s'agit d'une réaction de couplage croisé entre un électrophile R1X : dérivé halogéné vinylique ou arylique ou encore un dérivé sulfonylé (triflate) et un acide boronique R2B(OH)2 ou mieux, un ester boronique R2B(OR')2 ou encore un trialkylborane avec :
R1 : alcényle, aryle, benzyle, allyle, alcynyle ;
X = halogène (I, Br), OTf (triflate), N2+ ;
R2 : phényle, alcényle.
Un complexe du palladium tel que le Tétrakis (triphénylphosphine)palladium (0) : Pd(PPh3)4, est utilisé en quantité catalytique.
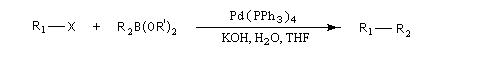
Le mécanisme simplifié est représenté ci-dessous.
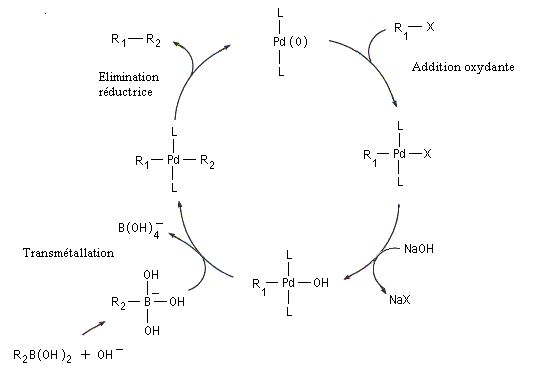
Il comporte les étapes suivantes :
L’activation par une base de l’entité boronique facilite l'étape de transmétallation. L'organoborane est habituellement préparé par hydroboration d'un alcyne ou par hydroboration d'un alcène.
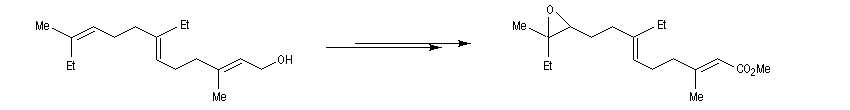
Un très gros avantage de ce type de réaction est que les réactifs peuvent posséder des groupes fonctionnels variés. Il est ainsi possible de coupler entre-elles des chaînes carbonées fonctionnalisées de grandes tailles lors d'une synthèse. Dans l'exemple suivant, l'une des chaînes de l'organoborane comporte un époxyde pourtant réputé comme étant particulièrement réactif.
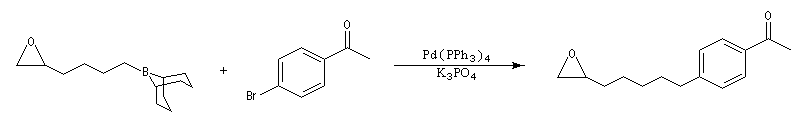
Le couplage de Suzuki a permis une synthèse élégante du bombykol [51]
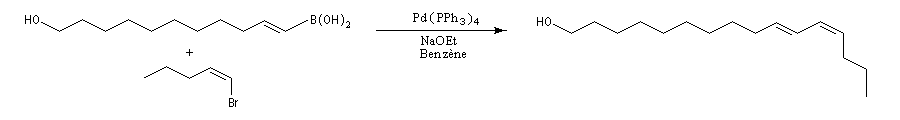
Ainsi que celle du rétinol (vitamine A1.)
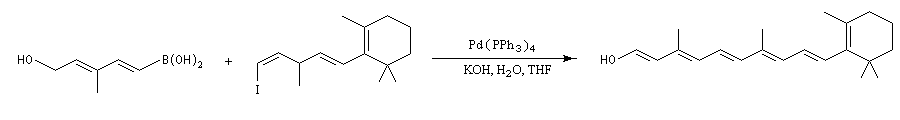
La réaction a été utilisée avec succès pour synthétiser de nombreuses structures complexes.
La dernière étape de la synthèse de la myxalamide A par C.H. Heathcock est une réaction de couplage de Suzuki entre un vinylborane de stéréochimie E et un iodotriene fonctionnalisé de stéréochimie Z [52]
Le coût assez modique des réactifs de départ permet une utilisation à l'échelle industrielle.
Alkylations de composés organométalliques
Réaction de Corey-House-Posner-Whitesides
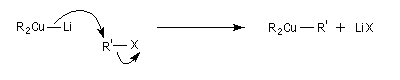
Cette réaction a permis à Corey et à ses collaborateurs de réaliser la synthèse de l'hormone juvénile Cecropia (1968).
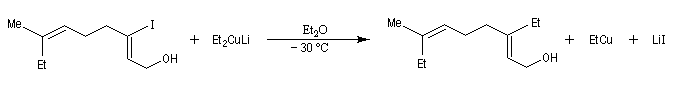
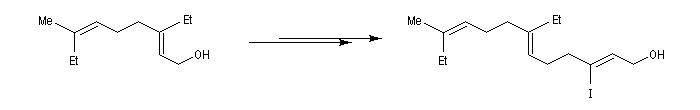
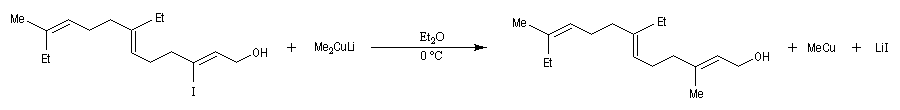
Avec les dérivés halogénés secondaires ou tertiaires l'élimination devient prépondérante.
Alkylation des alcénylcuprates
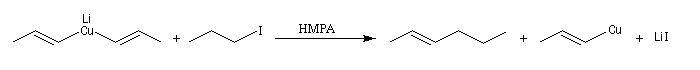
La stéréochimie de l'alcène est donc parfaitement contrôlée. Le mécanisme est de type SN2. Pour un exemple d'utilisation des cuprates vinyliques [35].
Les cuprates de Lipshutz permettent l'alkylation de dérivés halogénés secondaires.
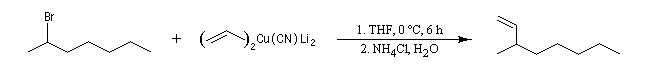
Réaction historique de Wurtz
La réaction repose sur la formation d'un organosodique.
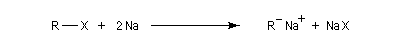
Le carbanion formé provoque une substitution nucléophile sur le dérivé halogéné.
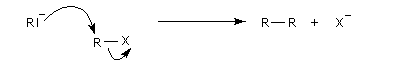
Un exemple original qui n'est guère généralisable est la préparation du bicyclo[1,1,0]butane [27].
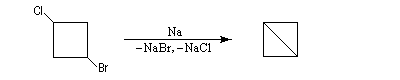
Le couplage de Wurtz est une réaction secondaire génante au cours de la préparation des composés organométalliques. En dehors de quelques cas très particuliers, ce type d'alkylation par les organosodiques n'est pas utilisable en synthèse car les carbanions étant des bases très puissantes, ils conduisent fréquemment à des réactions secondaires indésirables.
Réduction par les métaux
La réaction entre un dérivé halogéné et le zinc conduit à un organozincique. Ce dernier est ensuite traité par un donneur de proton comme l'acide acétique.
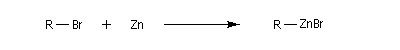
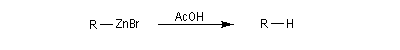
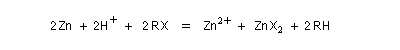
La réaction entre le zinc et le chlorure de triphénylméthyle conduit à la formation d'un radical organique relativement stable, le radical triphénylméthyle.
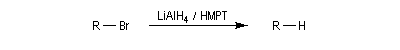
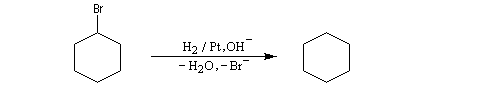
Bibliographie
Ouvrages et articles expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[13] J. F. Bunnett, Angew. Chem. Int. Ed. Engl. 1, 225 (1962).
[14] Sir Christopher Ingold- A Major Prophet of Organic Chemistry by Kenneth T. Leffek, JCE, June 1997
Vol. 74 No. 6 p. 625
[15] Hine J, J. Am. Chem. Soc. 1950, 72, 2438.
[16] Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides, R. F. Heck, J. P. Nolley, J. Org. Chem., 1972, 37 (14), pp 2320–2322
[49] Posner, G. H. Substitution Reactions using Organo Copper Reagents, Organic Reactions 1975, 22, 253.
[50] Anthony O. King, Nobuhisa Okukado and Ei-ichi Negishi
J. Chem. Soc., Chem. Commun., 1977, 683-684
[51] Miyaura, N.; Suzuki, A. Chem. Commun. 1979, 866.
[52] Mapp, A.K., Heathcock, C. H. Total Synthesis Myxalamide A.J. Org. chem. 1999, 64,23-27.
[17] Nucleophilic substitution
[18] Chemistry Comes Alive, nucleophilic substitution
[19] The Hard Soft Acide Base Principle
[20] Tyrian purple
[21] Cours sur les dérivés halogénés
[22] n-hexadecane by P. A. Levene
[23] linoleic acid by J. W. McCutcheon
[25] Benzocyclopropene by W. E. Billups, A. J. Blakeney, and W. Y. Chow
[26] 1,6-methano-10-annulene by E. Vogel, W. Klug, and A. Breuer
[27] Bicyclo[1,1,0]butane by Gary M. Lampman and James C. Aumiller
[28] Rearrangements induced by cationic or electron deficient sites by William Reusch
[29] Cyclopropanecarboxylic acid by Chester M. McCloskey and George H. Coleman
[30] Glossary of Terms used in Physical Organic Chemistry IUPAC Recommendations 1994
[31] Diphenylacetylene by Lee Irvin Smith and M. M. Falkof
[32] 1,1-dichloro-2,2-difluoroéthylène by J. C. Sauer
[33] Allène by H. N. Cripps and E. F. Kiefer
[34] Cyclobutene by J. Salaün and A. Fadel.
[35] (Z)-1-iodohexene by A. Alexakis, G. Cahiez, and J. F. Normant.
[36] Prix Nobel de chimie 2010
[37] Thèse P. E. Broutin.
[38] Thèse L. Chahen.
[39] Synthesis by metal mediated Coupling Reactions
[40] Heck reaction: Mechanism and it’s application in total synthesis of Natural Products by Venugopal Rao
A. Streitwieser Jr, C.H. Heathcock - Introduction to Organic Chemistry (Macmillan Publishing Co).
J. March - Advanced organic chemistry (Wiley Interscience 2006).
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, (Plenum Press, 1990).
N. T. Ahn - Introduction à la chimie moléculaire, (Ellipses, 1994).
C. Reichardt - Effets de solvants en chimie organique, (Flammarion Sciences, 1971).
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique (Ellipses, 1993).
P. Atkins - General Chemistry (Scientific American Books W.H Freeman and Co).
Metal-catalyzed Cross-coupling Reactions, Edited by F. Diederich and P. J. Stang, Wiley-VCH, New York, 2004
Organometallics in Synthesis. A manual, Schlosser, M., Second Edition, Wiley, New
York, 2002.
Pour aller plus loin
Suzuki cross coupling
Synthèse organique par voie organométallique par T. Ollevier
Organometallic compound of Lithium Sodium and Potassium by Michael K. Denk.
Organometallic chemistry by Worawan Bhanthumnavin
Bien qu'étant assez anciens, les ouvrages suivants conservent de l'intérêt :
C. K. Ingold - Structure and Mechanism in Organic Chemistry, Cornell Univ. Press,
Ithaca (1953).
J. Hine, Physical Organic Chemistry, 2nd Edition, McGraw-Hill, New York (1962).
R. Breslow - Mécanismes des réactions organiques (Edisciences, - 1970).
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Merci, dans ce cas, d'avoir la gentillesse de citer le nom de l'auteur afin d'éviter des conflits d'antériorité.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
décembre 2011



![1-bromobicyclo[2,2,2]octane](images24/quinubr.gif)