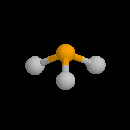
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Les alcools
Introduction
La chaîne principale est la chaîne la plus longue qui porte le groupe -OH. La numérotation de la chaîne est choisie de façon que le groupe -OH ait le numéro le plus petit. Le nom de l’alcool est formé en ajoutant le suffixe ol au nom de l’hydrocarbure possédant le même nombre d’atomes de carbone que la chaîne principale.
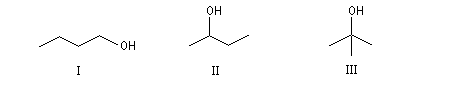
Les trois alcools suivants ont pour formule C4H10O. Ce sont des isomères de position.
|
I |
II |
III |
|
Butan-1-ol |
Butan-2-ol |
2-Méthylpropan-2-ol |
Notons qu'il existe deux molécules de butan-2-ol énantiomères.
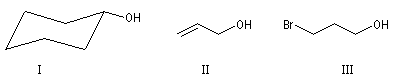
La chaîne carbonée peut être insaturée, si l’insaturation n’affecte pas l’atome de carbone portant le groupe hydroxyle.
|
I |
II |
III |
|
Cyclohexanol |
Prop-2-ène-1-ol |
3-Bromopropan-1-ol |
Les énols, composés dans lesquels le groupe -OH est lié à un atome de carbone insaturé ou les phénols dans lesquels ce groupe est lié à un cycle aromatique, ne sont pas des alcools.
Classe
Selon que l'atome de carbone portant le groupe caractéristique -OH est lié à 1, 2, 3 atomes de carbone, l'alcool est qualifié de primaire, secondaire, tertiaire. Le butan-1-ol, le butan-2-ol, le 2-méthylpropan-2-ol sont des isomères de position de formule brute C4H10O appartenant aux trois classes.
|
Alcool |
butan-1-ol |
butan-2-ol |
2-méthylpropan-2-ol |
|
Classe |
I |
II |
III |
Etat naturel
Beaucoup d'alcools existent à l'état naturel. Le méthanol était obtenu autrefois par distillation du bois. L'éthanol se forme par fermentation des jus sucrés. Le (10E, 12Z)-hexadéca-10,12-diène-1-ol ou bombykol est une phéromone sexuelle du bombyx qui a été isolée en 1959 par J. Butenandt.
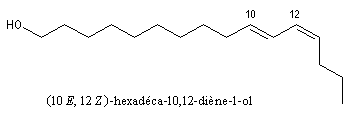
Le rétinol ou vitamine A est le précurseur biochimique des rétinoïdes qui jouent un rôle essentiel dans le mécanisme de la vision.
Le menthol est l'un des constituants de l'essence de menthe. Le cholestérol est le représentant le plus connu d'une famille de composés extrêmement importants en biochimie : les stéroïdes.
|
|
Le (E)-3,7-diméthyl-2,6-octadién-1-ol ou géraniol est un alcool terpénique présent dans l'essence de géranium.
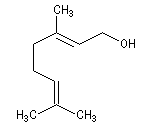 Le (Z)-3,7-diméthyl-2,6-octadién-1-ol, diastéréoisomère du précédent est le nérol.
Le (Z)-3,7-diméthyl-2,6-octadién-1-ol, diastéréoisomère du précédent est le nérol.
|
Méthanol CH3OH
C'est l'alcool dont le tonnage produit est le plus grand.
|
|
Le méthanol est produit par addition entre H2 et CO.
Les produits dérivés sont les suivants : 50 % méthanal, 10 % diméthyltéréphtalate (fibres polyesters), 10 % méthyltertiobutyléther MTBE (additif pour carburant), 6 % acide éthanoïque (par carbonylation avec CO), 13 % divers (méthylamine, chlorométhane, méthylméthacrylate). |
|
|
L'éthanol peut être obtenu par fermentation de sucres. Une autre voie d'accès est la synthèse à partir de l'éthène qui représente 30 % de la production en Europe et 60 % aux USA.
. Il est utilisé en tant que solvant, pour la synthèse de dérivés halogénés et d'éthanoate d'éthyle. |
|
|
Le cyclohexanol est obtenu par hydrogénation du phénol.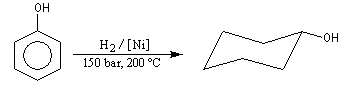 Son oxydation en acide adipique (hexanedioïque) est une étape de la synthèse du Nylon 6-6. |
Les polyols ou polyalcools sont des composés comportant plusieurs groupes -OH. L'éthane-1, 2- diol est utilisé dans la préparation de polyesters. Le propane-1, 2, 3-triol (glycérol) sert à la préparation de la nitroglycérine.
Caractéristiques géométriques et énergétiques
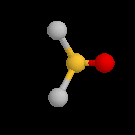
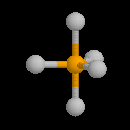
Par définition, l'atome de carbone fonctionnel est lié par des liaisons simples qui se développent selon les directions d'un tétraèdre. L'atome d'oxygène contracte deux liaisons simples respectivement avec l'atome de carbone et l'atome d'oxygène. La géométrie localement plane autour de l'atome d'oxygène dérive d'un arrangement tétraédrique des paires d'électrons. Puisque les paires non liantes occupent en moyenne un volume plus grand que les paires liantes, on prévoit un angle entre les liaisons a < 109°. Le tableau suivant regroupe quelques valeurs moyennes de grandeurs géométriques et énergétiques.
|
d (C-O) (nm) |
d (O-H) (nm) |
a (COH) (°) |
Do (C-O) (kJ.mol-1) |
Do (O-H) (kJ.mol-1) |
|
0,143 |
0,096 |
106 |
343 |
463 |
L'énergie de la liaison C-O est élevée. Sa réactivité s'explique avant tout par sa polarité et sa polarisabilité. La présence de l'atome d'oxygène plus électronégatif (3,5 dans l'échelle de Pauling) que les atomes de carbone (2,5) et d'hydrogène (2,1) ainsi que la géométrie de la molécule sont à l'origine d'un moment dipolaire permanent pour la molécule.
|
Alcool |
Constante diélectrique er |
Moment dipolaire m (D) |
|
Méthanol |
32,6 |
1,71 |
|
Ethanol |
24,3 |
1,68 |
Températures de changement d’état
Le tableau suivant regroupe les températures de changement d'état de quelques alcools courants.
|
Nom de l’alcool |
TF (°C) |
TE (°C) |
Densité d |
|
méthanol |
-97 |
64,7 |
0,792 |
|
éthanol |
-114 |
78,3 |
0,789 |
|
propan-1-ol |
-126 |
97,2 |
0,804 |
|
propan-2-ol |
-88 |
82,3 |
0,786 |
|
butan-1-ol |
-90 |
117,7 |
0,810 |
|
2-méthylpropan-2-ol |
2 |
82,5 |
0,789 |
|
hexan-1-ol |
-52 |
155,8 |
0,820 |
|
dodécanol |
24 |
259 |
0,831 |
Ces constantes physiques sont beaucoup plus élevées que celle des hydrocarbures de même masse molaire.
|
Composé |
propane (M = 44 g.mol-1) |
éthanol (M = 46 g.mol-1) |
|
Température d'ébullition |
- 42 °C |
78,5 °C |
Cela s'explique par l’association des molécules d'alcool par liaison hydrogène. Le dessin ci-dessous schématise un exemple d'association dans le cas du méthanol.
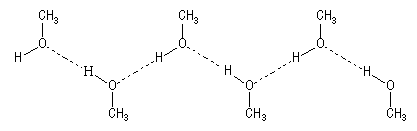
Les liaisons hydrogène se rencontrent chaque fois que l'atome d'hydrogène est lié à un atome fortement électronégatif (F, S, O). La taille très faible de l'atome d'hydrogène (rayon de Van der Waals : rW = 120 pm) lui permet d'approcher très près de l'atome d'oxygène et d'interagir fortement avec lui.
Les alcools en tant que solvants
Du fait de la présence du groupe -OH, les alcools jusqu'à 5 atomes de carbone sont très solubles dans l'eau avec laquelle ils s'associent par liaisons hydrogène. L'éthanol est miscible à l'eau en toute proportions. Le mélange n'a pas un caractère idéal et il s'effectue avec contraction de volume et dégagement de chaleur. Notons qu'il est impossible de préparer l'alcool absolu (100 % en éthanol) par distillation du mélange éthanol-eau car il existe un azéotrope positif (à point d'ébullition minimum) pour une teneur de 95 % en alcool.
L'éthanol et le méthanol dissolvent également assez bien certains composés ioniques. Comme ils sont miscibles à de nombreux composés organiques on les utilise fréquemment en synthèse organique comme solvants, par exemple dans des réactions de substitutions ou le nucléophile est un ion halogénure.
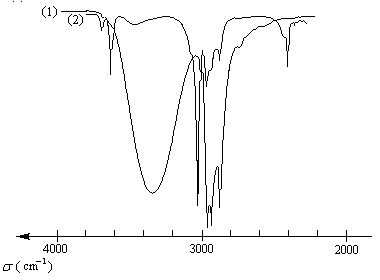
Spectroscopie infrarouge
Le spectre suivant est celui de l'hexan-1-ol. Il est typique du spectre infrarouge d'un alcool pur.
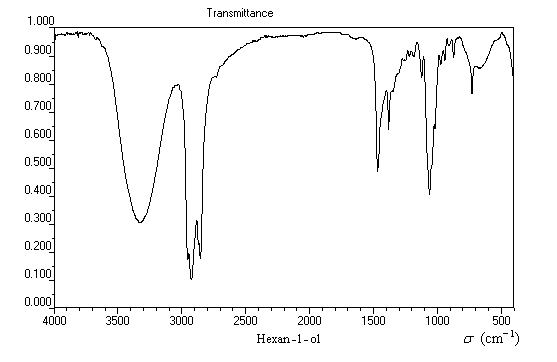
|
|
Influence de la nature du solvant sur le nombre d'onde de la vibration de valence de la liaison O-H :
|
On notera que l'association par liaison hydrogène abaisse le nombre d'onde d'absorption du vibrateur O-H. On peut s'en souvenir en remarquant que l'atome d'hydrogène étant impliqué à la fois dans la liaison hydrogène et dans une liaison avec l'atome d'oxygène, cette dernière subit un certain relâchement.
Liaisons hydrogène intramoléculaires : dans certaines molécules comme celles de polyols on observe des liaisons hydrogène intramoléculaires. Il est facile de distinguer les liaisons intermoléculaires et les liaisons intramoléculaires par spectroscopie infrarouge. Par dilution dans un solvant comme CCl4, la bande d'absorption due aux premières disparaît mais pas celle due aux secondes.
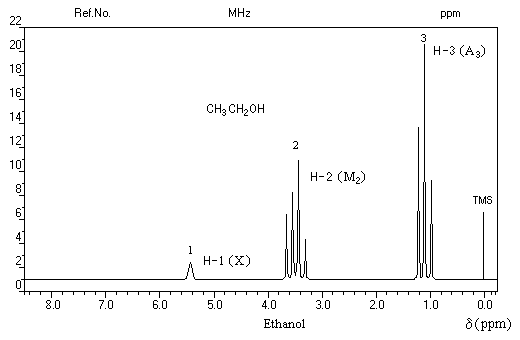
Spectroscopie de RMN
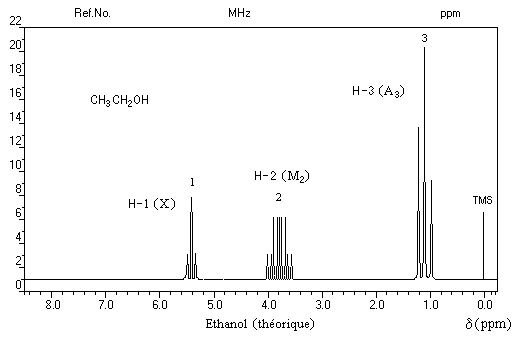
Le spectre de RMN de l'éthanol très pur (éthanol absolu) effectué à haute résolution présente trois groupes de protons de déplacements chimiques différents. C'est un système A3M2X. Les protons du méthylène -CH2 - (M2) sont couplés à la fois avec ceux du méthyle -CH3 (A3) et le proton du groupe -OH (X).
|
Groupe de protons |
CH3 (A3) |
CH2 (M2) |
H (X) |
|
Signal |
triplet |
quadruplet dédoublé |
triplet |
Le spectre de l'éthanol à 95 % (donc contenant 5 % d'eau) est plus simple. Les trois groupes de protons A, M, X sont toujours présents mais le signal du proton du groupe -OH (X) est un singulet élargi. Du fait de la mobilité de l'atome d'hydrogène du groupe hydroxyle, le couplage avec les autres protons disparaît.
|
Groupe de protons |
CH3 (A3) |
CH2 (M2) |
H (X) |
|
Signal |
triplet |
quadruplet |
singulet |
![]()
Notons qu'il est difficile d'attribuer un déplacement chimique précis à ce type de protons car la fréquence de résonance est fonction de la quantité d'acide introduite.
Une technique de simplification consiste à ajouter quelques gouttes d'eau lourde D2O. On observe alors l'équilibre :
![]()
Cela fait disparaître les pics dus aux protons échangeables car D ne résonne pas en RMN 1H dans le domaine de fréquence étudié.
En revanche on peut observer un signal dû au proton du groupe -OH sous forme de multiplet dans une structure possédant une liaison hydrogène intramoléculaire. L'échange du proton qui partage son affinité entre deux sites est suffisamment ralentie vis à vis du phénomène RMN pour que le couplage redevienne observable comme dans la structure ci-dessous :
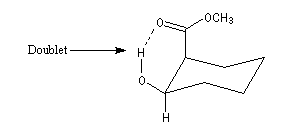
De telles informations sont extrêmement précieuses pour préciser la stéréochimie des molécules complexes.
Réactions d'oxydo-réduction
Réactifs usuels impliquant des éléments de transition
Les oxydants les plus classiques sont ceux qui impliquent des dérivés oxygénés de métaux de transition.
|
|
Le dichromate de potassium est un solide de couleur orange. Il est dissous dans une solution d'acide sulfurique. Le volume est complété par de l'eau distillée. Les composés du Cr (VI) sont dangereux. Il présentent malheureusement la propriété d'induire certains cancers. Comme l'absorption par voie cutanée constitue le risque principal, il est indispensable d'utiliser des gants pour manipuler ces composés. On peut effectuer le dosage de l'éthanol en le faisant réagir avec un volume connu de solution titrée de dichromate de potassium en excès ce qui permet de rendre la réaction d'oxydation quantitative. Le dichromate restant est réduit par une solution titrée de sel de Mohr. |
Influence de la classe de l'alcool
Intéressons-nous à l'oxydation de 3 des 4 alcools isomères de formule brute C4H10O appartenant à chaque classe : le butan-1-ol, le butan-2-ol et le 2-méthylpropan-2-ol.
|
|
Les deux tubes de gauche contiennent respectivement du butan-1-ol et une solution de dichromate de potassium dans l'acide sulfurique. Dans le tube de droite, on a introduit une petite quantité d'alcool dans la solution de dichromate de potassium. Une coloration bleu-vert se développe qui témoigne de la réduction des ions Cr2O72- en ions Cr3+. |
|
|
Dans le tube où s'est déroulée l'oxydation, on a ajouté une petite quantité de pentane. Après agitation, le butanal formé par oxydation de l'alcool se concentre dans cette phase organique (phase supérieure). Quelques mL de la phase organique surnageante sont ajoutés dans deux autres tubes :
|
Avec le butan-2-ol, la réaction est plus lente qu'avec le butan-1-ol. Le test à la 2,4-DNPH est positif. En revanche, on n'observe pas de réaction avec le réactif de Schiff. Avec le 2-méthylpropan-2-ol, il n'y a pas de réaction. Les résultats sont résumés dans le tableau suivant :
|
Alcool |
butan-1-ol |
butan-2-ol |
2-méthylpropan-2-ol |
|
Vitesse |
rapide à froid |
lente à froid |
- |
|
Produit |
butanal |
butanone |
- |
Les différences de comportement des alcools vis à vis de l'oxydation sont très nettes suivant la classe à laquelle ils appartiennent. La présence d'un atome d'hydrogène sur l'atome fonctionnel est indispensable pour que l'alcool soit oxydé. Les alcools tertiaires, ne sont pas oxydés.
|
Classe |
primaire |
secondaire |
tertiaire |
|
Produit d'oxydation |
aldéhyde |
cétone |
- |
Notons qu'un alcool tertiaire comme le 2-méthylpropan-2-ol est facilement déshydraté en alcène par chauffage modéré en présence d'acide. Cet alcène peut alors subir une coupure oxydante. C'est la raison pour laquelle les alcools tertaires donnent un test positif avec les oxydants forts en milieu acide.
Remarque : le 2-méthylpropan-2-ol est solide à la température ordinaire. Lorsqu'on veut réaliser des réactions tests avec cet alcool il ne faut pas utiliser les quelques gouttes de liquide obtenues en retournant la bouteille. En effet il peut s'agir des impuretés présentes dans le composé.
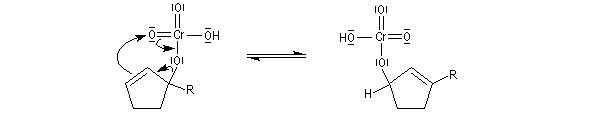
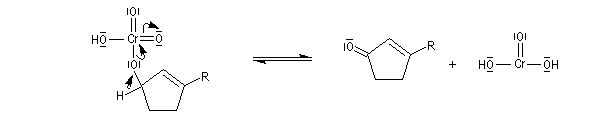
Le mécanisme de l'oxydation chromique des alcools a été étudié sur l'exemple de l'alcool isopropylique par Westheimer. Il y a formation réversible d'un ester chromique.
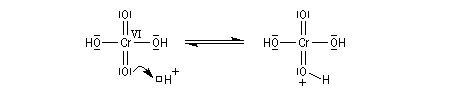
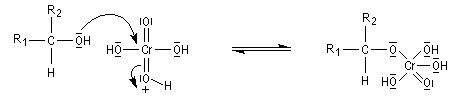
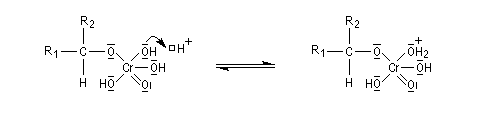
L'élimination d'un proton dans l'étape ci-dessous qui est cinétiquement déterminante, a été prouvée par l'existence d'un effet isotopique en utilisant un alcool deutéré.
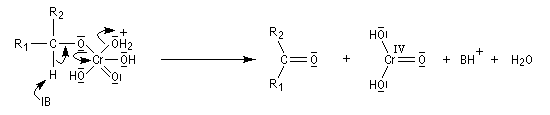
Alcools secondaires
L'oxydation des alcools secondaires conduit aux cétones. Le réactif de Jones est un réactif souvent employé (on dissout 26,72 g de CrO3 dans 23 mL de H2SO4 concentré puis on étend avec de l'eau jusqu'à un volume de 100 mL).
Ainsi, le cyclohexanol est aisèment oxydé en cyclohexanone par le réactif de Jones.
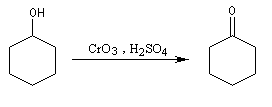
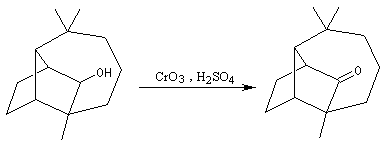
Alcools primaires
L'oxydation d'un alcool primaire conduit, dans un premier temps, à un aldéhyde. Mais les aldéhydes étant des réducteurs forts il faut prendre des précautions spéciales pour s'arrêter à ce stade. Un certain nombre de techniques sont utilisables.
Les aldéhydes sont généralement plus volatils que les alcools parents car ils ne forment pas de liaison hydrogène. Il est parfois possible de distiller l'aldéhyde au fur et à mesure de sa formation ce qui a pour effet de supprimer le contact avec l'oxydant.
On peut ainsi préparer le butanal par oxydation du butan-1-ol avec Na2Cr2O7 en présence d'acide sulfurique. Le rendement n'est cependant pas très bon.
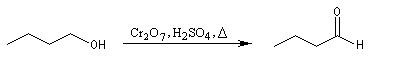
Une autre technique d'isolement est utilisée dans le test des alcools décrit plus haut. La méthode consiste à utiliser deux phases non miscibles : par exemple eau et pentane. Au fur et à mesure de sa formation, le butanal plus soluble dans le pentane que dans l'eau est extrait du milieu aqueux ce qui, là encore, évite le contact avec le réactif d'oxydation.
L'étude du mécanisme de la réaction d'oxydation des aldéhydes montre que celle-ci passe par la formation d'un ester chromique qui implique l'hydrate du carbonylé (composé d'addition entre le carbonylé et l'eau). L'idée est d'éviter la formation de cet hydrate en opérant en l'absence d'eau. Le chlorochromate de pyridinium PCC sur alumine ou le dichromate de pyridinium (PyNH+)2 Cr2O72- (PDC) sont des réactifs de choix dans de telles oxydations.
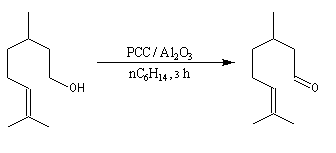
Avec des molécules complexes, possédant plusieurs fonctions, se pose le problème de la chimiosélectivité du réactif d'oxydation. Plusieurs réactifs ont été proposés pour résoudre ce problème :
|
|
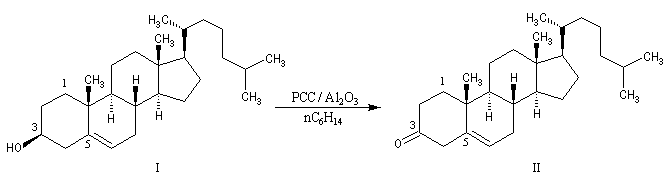
Le chlorochromate de pyridinium C5H5NH+ClCrO3- ou PCC, introduit par le chimiste américain E. J. Corey, est un réactif souvent utilisé (12 g de CrO3 dans 22 mL de HCl 6 M on ajoute 9,5 g de pyridine pendant 10 min en maintenant la température à 40 °C. On refroidit le mélange à 0 °C . Le produit cristallise. Il est séché sous vide pendant 1 h). PCC est également utilisé sur alumine (chauffer la solution précédente vers 40 °C jusqu'à ce que le solide se dissolve. Ajouter 100 g d'alumine en agitant. Evaporer le solvant avec un évaporateur rotatif. Sécher sous vide pendant 2 h à la température ambiante). L'avantage de PCC sur alumine tient à la grande facilité de récupération du produit d'oxydation [3]. |
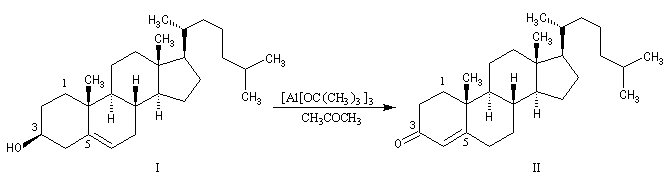
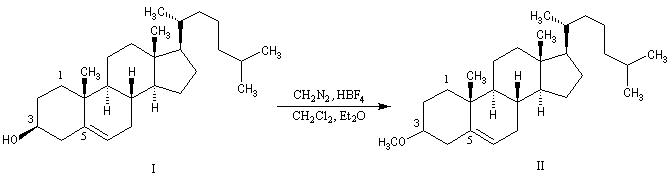
Au moyen du PCC, on peut réaliser l'oxydation chimiosélective du cholestérol (I) en cholest-5-ène-3-one (II) sans provoquer l'isomérisation de cette cétone. En revanche, l'oxydation d'Oppenauer du cholestérol fournit la cholest-4-ène-3-one, a-énone plus stable que son isomère précédent.
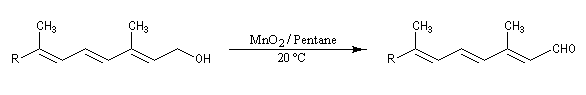
Oxydations des alcools allyliques
Les alcools allyliques et benzyliques sont plus réactifs que les alcools saturés (une manifestation de la mobilité protonique en position allylique). Ils sont oxydés par le dioxyde de manganèse MnO2 dans des conditions douces. On obtient des aldéhydes a, b-insaturés. A partir du rétinol (vitamine A) on peut ainsi obtenir le rétinal, un composé important dans la chimie de vision.
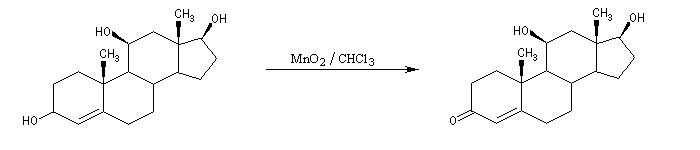
L'oxydation des alcools tertiaires allyliques est une réaction très utile en synthèse qui conduit à une a-énone après transposition du squelette carboné.
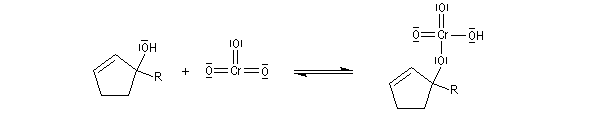
L'exemple ci dessous concerne la dernière étape de la synthèse de la jasmone par P. A. Grieco (1972).
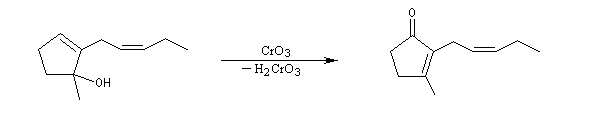
Oxydations en milieu biologique
En milieu biologique, l'oxydation des alcools fait intervenir des enzymes appelées alcool-déshydrogénases. Le transfert des équivalents réducteurs vers le substrat est assuré par des coenzymes dont le plus important est le système NAD+/NADH.
NAD+ peut fixer un ion hydrure c'est à dire l'équivalent d'un ion H+ et de 2 électrons. La demi-équation électronique s'écrit :
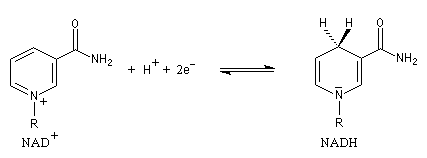
Le potentiel standard (conditions biologiques : T = 37 °C) de ce système est : E0' = -0,32 V.
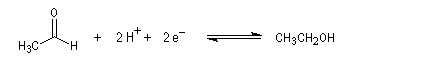
Pour le système éthanal/éthanol, on a : E0' = -0,20 V.
Notons que l'oxydation d'un alcool nécessite une base pour arracher un atome d'hydrogène de l'alcool.
L'oxydation d'un alcool chiral par NAD+ est une réaction énantiosélective. Dans l'oxydation du deutérioéthanol de configuration absolue S par NAD+ on obtient de l'éthanal deutérié tandis que l'atome d'hydrogène est retrouvé dans NADH.
Inversement, en présence d'une enzyme spécifique, NADH est capable de transférer l'atome d'hydrogène prochiral pro-R sur la face prochirale re de l'éthanal.
|
|
Le nicotinamide adénine dinucléotide est un coenzyme de type soluble. Il est fixé à l'enzyme pendant la réaction puis est libéré. Une seconde réaction indépendante régénère le coenzyme.
Notons que NAD+ est un composé aromatique. En revanche NADH ne l'est pas. Le passage de NAD+ à NADH correspond donc à une diminution de la stabilité du système. |
![]()
70 % de la production de méthanal utilise ce procédé.
L'oxydation de l'éthanol avec le cuivre comme catalyseur conduit à l'éthanal.
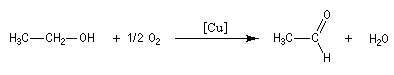
Oxydations avec coupure de la chaîne carbonée
Dans des conditions assez vigoureuses, les alcools secondaires cycliques sont oxydés en cétones qui s'oxydent à leur tour avec rupture de la chaîne carbonée. L'oxydation de la cétone s'effectue par l'intermédiaire de la forme tautomère énol. L'oxydation du cyclohexanol par l'acide nitrique, permet la synthèse de l'acide hexane-1,6-dioïque encore appelé acide adipique [3].
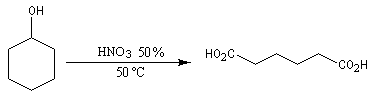
L'acide adipique est l'un des réactifs utilisés dans la synthèse du Nylon 6-6.
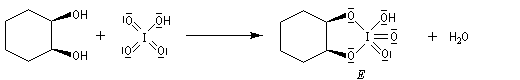
Coupure des a-glycols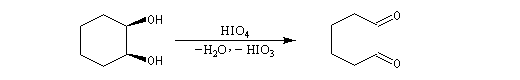
Elle fait intervenir un ester de l'acide périodique comme intermédiaire.
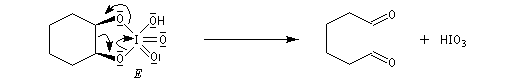
Un autre exemple concerne la coupure de l'éthane-1,2-diol (éthylène glycol).
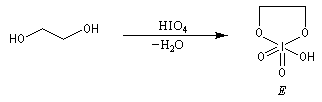
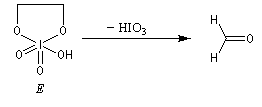
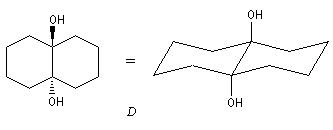
La réaction ci-dessous est une étape de la synthèse du cholestérol (Woodward 1952).
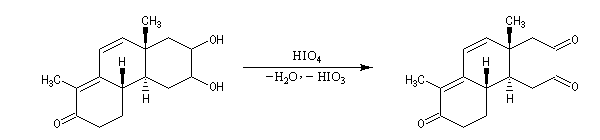
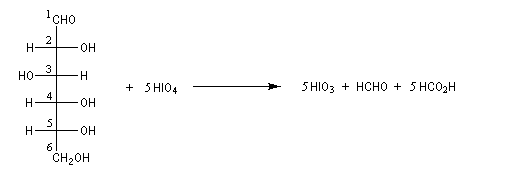
Le tétracétate de plomb Pb(OAc)4 est aussi un oxydant efficace dans le clivage des a-glycols (réaction de Criegee) [37].
Oxydations utilisant le DMSO activé
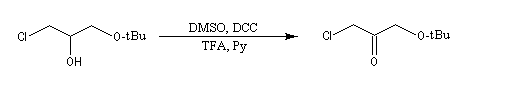
Plus récemment D. Swern a utilisé le DMSO activé par différents réactifs, notamment le chlorure d'oxalyle (chlorure d'acyle de l'acide oxalique) [29]. Notons que cette réaction doit être effectuée en suivant un protocole opératoire précis, compte-tenu du risque d'explosion qu'elle peut présenter si la température n'est pas
contrôlée.
La réaction d'oxydation d'un alcool comporte les étapes suivantes :
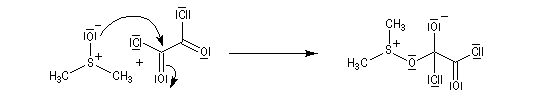
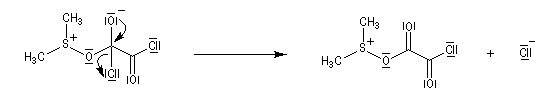
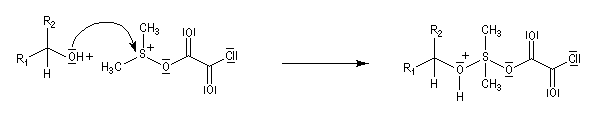
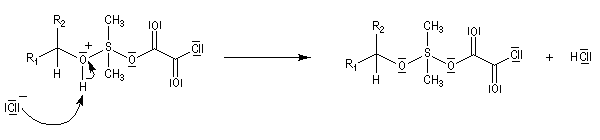
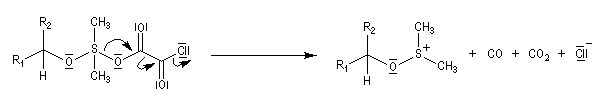
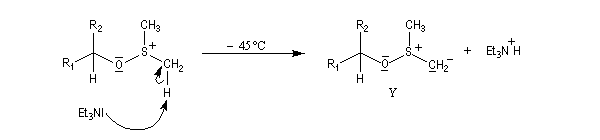
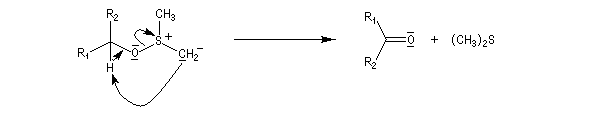
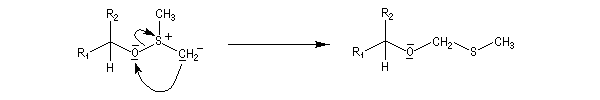
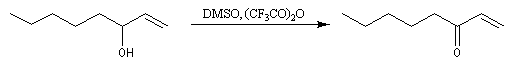
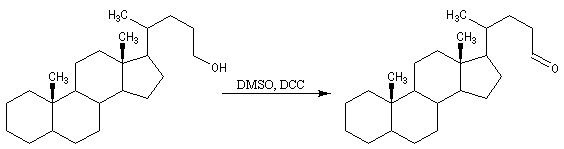
Il existe plusieurs variantes de l'oxydation de Swern. Dans l'oxydation de Parikh-Doering, le DMSO est activé par un mélange de SO3 et de pyridine [35].
Oxydation de Corey-Kim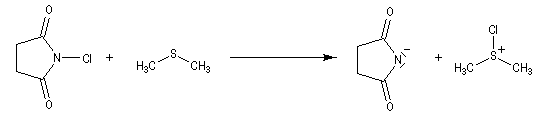
L'alcool réagit avec le soufre déficient en électrons.
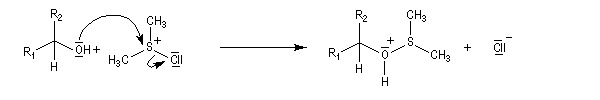
La suite ressemble à l'oxydation de Swern.
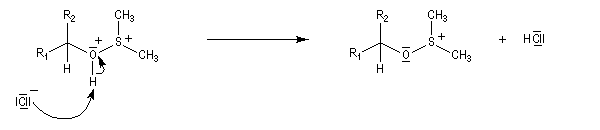
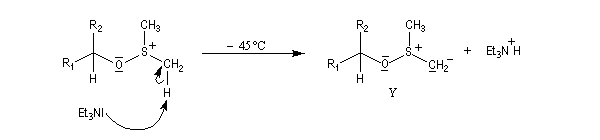
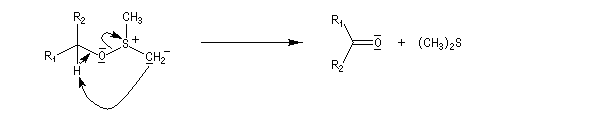
Voici un exemple d'application.
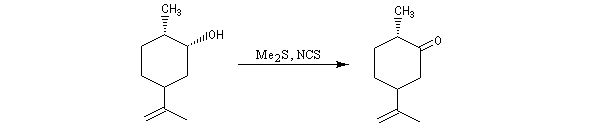
![]()
Le mécanisme fait intervenir un état de transition cyclique.
Utilisation du réactif de Dess-Martin
Le réactif de Dess-Martin est un oxydant des alcools primaires et secondaires. Sa préparation s'effectue en deux étapes [25] :
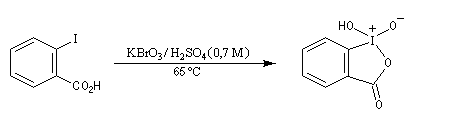
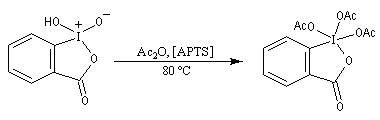
Le réactif de Dess-Martin est utilisé comme oxydant des alcools primaires (et dans certains cas secondaires).
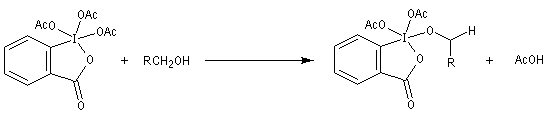
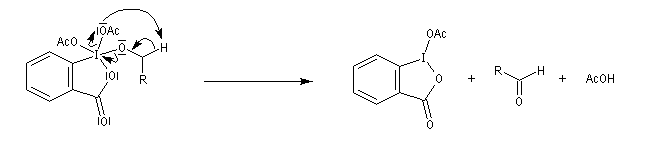
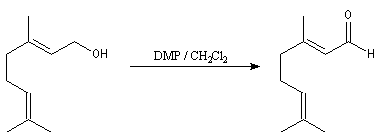
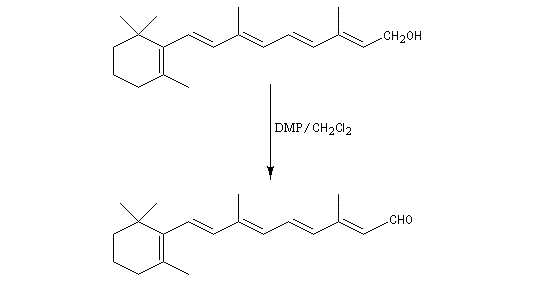
Un autre exemple est la synthèse du rétinal à partir du rétinol.
Propriétés acido-basiques
Propriétés acides
Les alcools ne manifestent aucune propriétés acido-basiques dans l'eau. Dans ce solvant, l'ionisation de la liaison OH d'un alcool comme le méthanol est extrêmement faible. Ainsi la constante thermodynamique de la réaction suivante vaut : K = 10-16 à 25 °C.
![]()
Puisque le produit ionique de l'eau est Ke = 10-14 à cette température, la constante thermodynamique de la réaction entre cet alcool et l'hydroxyde de sodium a pour valeur : K = 10-2.
![]()
L'ion hydroxyde ne peut donc produire l'ion méthanolate (et a fortiori les autres ions alcanolate) qu'en quantité très faible. Inversement, les ions alcanolate sont des bases fortes nivelées.
|
|
Les ions alcanolate sont des bases fortes nivelées par l'eau en ions hydroxyde OH-. L'ajout de quelques gouttes d'éthanolate de sodium dans une solution aqueuse de phénolphtaléine provoque l'apparition d'une coloration rose qui témoigne de la présence d'ions HO-.
|
Dans l'eau, les autres alcools sont moins acides que le méthanol. Les pKa des couples acido-basiques sont mesurés dans des solvants non aqueux puis extrapolés en phase aqueuse. Les valeurs suivantes sont donc approximatives :
|
Alcool |
CH3OH |
C2H5OH |
(CH3)2CHOH |
(CH3)3COH |
|
pKa (ROH/RO-) |
16 |
18 |
18 |
19 |
En revanche, dans les solvants dipolaires aprotiques comme le DMSO les différences d'acidité ont tendance à s'estomper.
A propos de l'acidité des alcools : la rationalisation de la différence d'acidité des alcools appartenant à différentes classes a déjà fait couler beaucoup d'encre. Longtemps attribuée à l'effet inductif donneur des groupes alkyles, la basicité plus grande de l'ion tertiobutylate par rapport à l'ion méthylate est due, pour une bonne part, à la solvatation moins importante de cette base dans les solvants protiques (schématiquement, plus un ion est volumineux, moins il est solvaté). Cela a été notamment montré par R. Mc Iver (Université de Californie Irvine) dans les années 70, en utilisant une technique spéciale de spectrométrie de masse (la résonance cyclotronique ionique) qui permet d'étudier les réactions chimiques en l'absence de solvatation.
Le couple menthol-ion mentholate a été souvent utilisé pour déterminer les pKa des couples acido-basiques comme alternative aux méthodes électrochimiques car les pouvoirs rotatoires spécifiques de l'alcool et de son sel sont très différents.
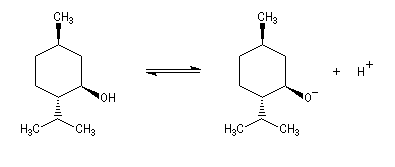
Plusieurs méthodes sont mises en œuvre pour déprotoner quantitativement les alcools.
|
Couple |
H2/H- |
NH3/NH2- |
|
pKa |
35 |
38 |
![]()
Si l'on se réfère à l'élément hydrogène il s'agit d'une médiamutation.
![]()
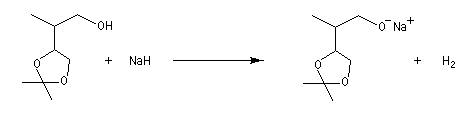
Avec les alcools primaires, les plus faciles à déprotoner, on utilise le sodium :
E0 (Na+/Na) = -2,7 V. Avec les alcools tertiaires comme le tertiobutanol qui sont moins réactifs on utilise le potassium.
Propriétés basiques
L'atome d'oxygène des alcools manifeste des propriétés basiques. Les alcools peuvent être protonés en présence d'un acide fort comme l'acide sulfurique.
|
Alcool |
CH3OH |
CH3CH2OH |
(CH3)2COH |
|
pKa |
-2,2 |
-2,4 |
-3,8 |
Ces réactions sont surtout importantes lorsqu'elles précédent le départ d'eau en tant que nucléofuge.
Propriétés nucléophiles de l'oxygène
Synthèse de Williamson des éthers
L'atome d'oxygène des alcools n'est pas suffisamment nucléophile pour déplacer directement des nucléofuges moyens. Un moyen d'exalter la réactivité nucléophile de l'oxygène est d'utiliser l'ion alcoolate.
La synthèse A. W. Williamson des éthers est basée sur la réaction de substitution nucléophile entre un alcoolate et un halogénure. La nucléophilie de l'alcoolate est exaltée par l'utilisation d'un solvant dipolaire aprotique comme le DMSO. Il s'agit d'une substitution nucléophile bimoléculaire. Ainsi, la méthode est particulièrement efficace lorsque le substrat est un halogénure primaire.
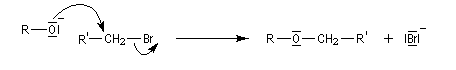
On peut, par cette méthode, synthétiser des éthers dissymétriques. L'exemple ci-dessous concerne la préparation de l'éther méthylique du menthol.
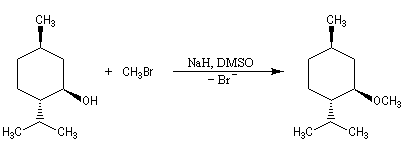
Comme les alcoolates sont à la fois nucléophiles et basiques. Le rendement de la réaction chute quand la classe du dérivé halogéné augmente du fait de la compétition avec la réaction d'élimination. Avec un substrat tertiaire l'élimination devient quasi-exclusive. La synthèse du méthyltertiobutyléther (MTBE) est possible en utilisant comme substrat le bromométhane et comme réactif l'ion 2-méthylpropanolate.
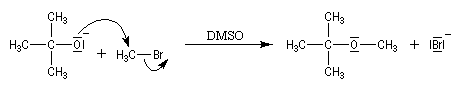
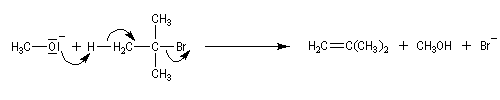
En revanche, avec le 2-bromo-2-méthylpropane comme substrat et l'ion méthanolate comme réactif, le rendement en éther est voisin de zéro car la réaction d'élimination l'emporte.
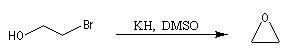
Cette méthode permet la synthèse des époxydes (oxacyclopropanes) en utilisant des halohydrines comme composés de départ. La réaction est une SN intramoléculaire.
Lorsqu'on effectue ce type de réaction à partir du (2R, 3R)-2-hydroxy-3-bromobutane, on obtient un époxyde méso. Cela montre que l'atome d'oxygène et le nucléofuge sont en position anticoplanaire lors de la substitution.
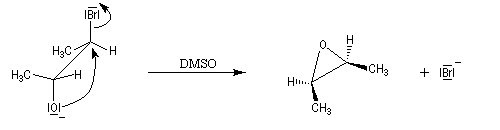
En série cyclohexanique, la formation d'époxyde est possible à condition que l'ion alcoolate soit en position anticoplanaire par rapport au nucléofuge. Il faut donc utiliser un dérivé trans.
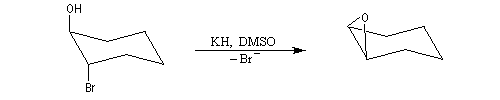
Dans le cas du dérivé cis, la substitution intramoléculaire est impossible. L'équilibre conformationnel place un atome d'hydrogène en position anticoplanaire par rapport à l'atome de brome.
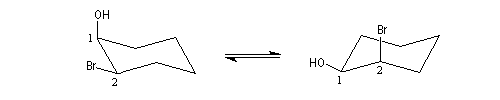
L'élimination devient possible. On obtient un énol qui se tautomérise en cétone.
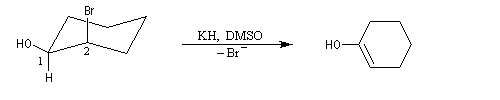
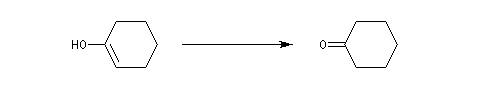
Les époxydes sont des composés importants car ils peuvent être ouverts par un grand nombre de réactifs nucléophiles (RMgX, RO-, LiAlH4, OH-, ...)
Par une méthode analogue, on sait synthétiser des éthers cycliques à 3, 4, 5, 6, 7 chaînons. Les meilleurs rendements sont obtenus pour les cycles à 3, 5 et 6 chaînons. Plusieurs facteurs interviennent pour déterminer ce pourcentage :
Une substitution intramoléculaire de ce type intervient dans la dernière étape de la synthèse de Darzens des époxyesters.
Alcoolyse des halogénures tertiaires
La synthèse des éthers dérivant de dérivés halogénés tertiaires est néanmoins possible par un mécanisme monomoléculaire SN1 s'il peut se former un carbocation relativement stable. Le nucléophile est alors moins puissant puisque c'est simplement l'alcool.
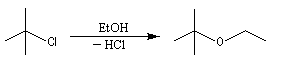
Acétalisation
Hydroxyacétalisation
La réaction entre un alcool et un aldéhyde conduit à un hydroxyacétal. Avec une cétone on obtient un hydroxycétal. La transformation conduit à un équilibre pour lequel les produits sont défavorisés.
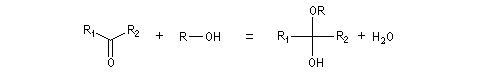
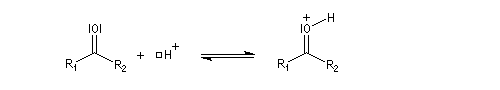
Cette réaction est l'objet d'une catalyse acido-basique généralisée. En milieu acide, utilise souvent l'acide paratoluène sulfonique (APTS) de préférence à l'acide sulfurique car il n'est pas oxydant.
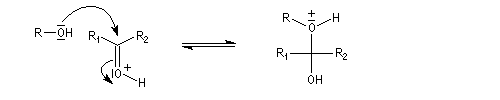
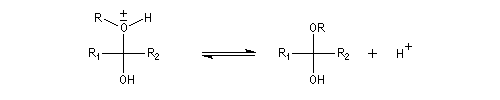
En milieu basique, une petite quantité d'alcool est déprotonée.
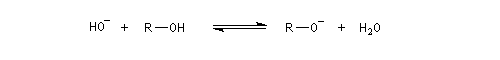
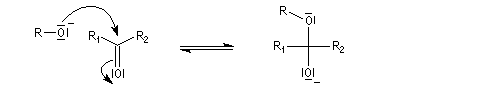
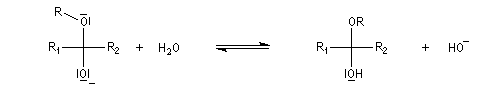
Les hémiacétalisations conduisant à des cycles à 5 ou 6 chaînons sont thermodynamiquement favorisées grâce à l'effet entropique.
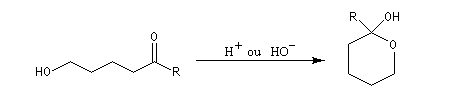
Hémiacétalisation des sucres
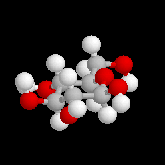
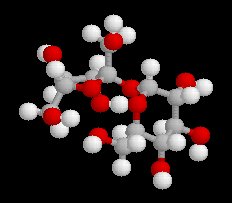
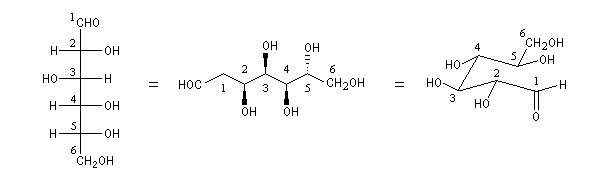
Le glucose naturel est le (2R, 3S, 4R, 5R)- 2, 3, 4, 5, 6-pentahydroxyhexanal. Il est représenté ci-dessous en projection de Fischer et en utilisant le mode de représentation de Cram.
L'hémiacétalisation intramoléculaire des sucres comme le glucose fournit des cycles à 6 chaînons appelés pyranoses ou à 5 chaînons appelés furanoses. Dans le cas du glucose, la formation de cycles à 6 chaînons est de loin la plus importante. Nous allons nous intéresser à ce dernier cas. La réaction conduit à un équilibre dont la position est en faveur du produit comme on l'a vu plus haut. Puisque la réaction peut affecter l'une ou l'autre des deux faces prochirales du groupe carbonyle, on obtient deux produits diastéréo-isomères.
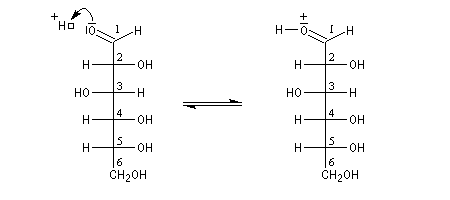
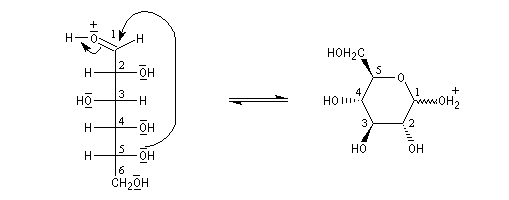
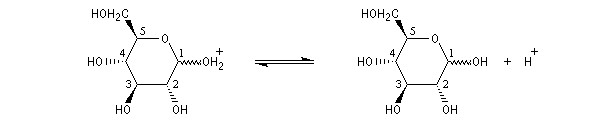
Ces diastéréo-isomères diffèrent par la configuration absolue d'un seul atome de carbone asymétrique. De tels diastéréo-isomères sont appelés des épimères. Pour distinguer ces épimères particuliers dont le carbone asymétrique est le carbone hémiacétalique et non un atome de carbone quelconque, on leur donne le nom d'anomères. Le mot anomère est formé à partir des mots grecs ano, en tête, et meros, partie.
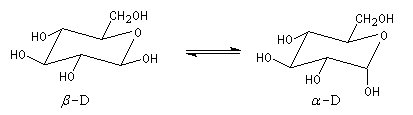
|
|
|
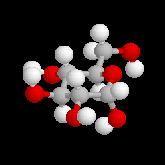
Le b-D-glucopyranose est représenté à gauche et le a-D-glucopyranose est représenté à droite. |
La stabilité relative des anomères a et b fait intervenir plusieurs facteurs antagonistes :
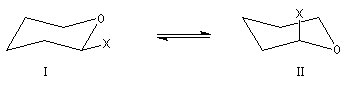
La solution est le siège d'un équilibre qui s'établit par l'intermédiaire de la forme ouverte. L'équilibre d'épimérisation s'accompagne d'un phénomène optique appelé mutarotation. Lorsqu'on dissout dans l'eau l'un des deux composés purs, le pouvoir rotatoire du mélange passe de la valeur caractéristique de ce composé à une valeur correspondant au mélange en équilibre des deux anomères.
|
Composé |
a -D-glucopyranose |
b -D-glucopyranose |
Mélange à l'équilibre |
|
[a]D (°.g-1.cm3.dm-1) |
112 |
19 |
52,2 |
La mesure du pouvoir rotatoire du mélange permet alors de déterminer les concentrations des anomères à l'équilibre. En effet, soit x, la fraction molaire en anomère a, en faisant l'hypothèse que la loi d'additivité est applicable (interactions négligeables entre les anomères) et que le pourcentage de la forme ouverte est très faible, on aura :
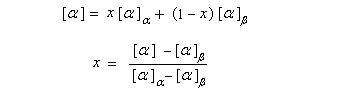
|
Composé |
a -D-glucopyranose |
b -D-glucopyranose |
|
x (équilibre) |
0,36 |
0,64 |
Expérimentalement, on constate donc que c'est l'anomère b qui est majoritaire à l'équilibre.
Synthèse des acétals et des cétals
Les hydroxyacétals et les hydroxycétals peuvent réagir avec un équivalent d'alcool pour donner respectivement des acétals et des cétals. La transformation conduit à un équilibre.
Comme on l'a vu précédemment, la formation des hydroxyacétals et des hydroxycétals est l'objet d'une catalyse acido-basique générale. En revanche, la synthèse des acétals est catalysée spécifiquement par les acides.
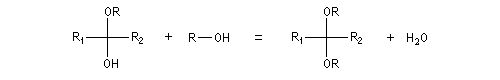
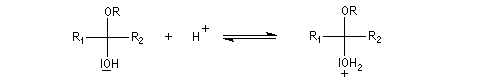
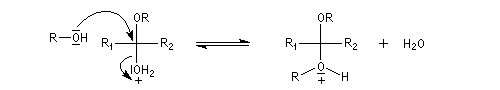
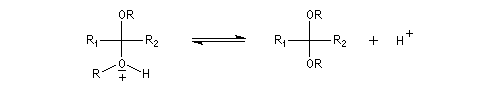
L'équilibre est défavorable au produit. Un moyen de déplacer sa position consiste à éliminer l'eau. On y parvient en ajoutant du toluène au mélange. L'eau et le toluène forment un hétéroazéotrope. On piège l'eau dans un décanteur du type Dean-Stark. Les acétals et les cétals sont, comme les éthers, des composés peu réactifs. Ils sont stables en milieu basique mais, en milieu acide, en présence d'un excès d'eau ils redonnent facilement les composés parents. Cette particularité permet de les utiliser comme groupes protecteurs des composés carbonylés ou des alcools.
|
|
Le saccharose est un diholoside formé par la réunion de deux oses : le glucose et le fructose. Les deux hydroxydes hémiacétaliques de ces oses forment une fonction acétal. La molécule est stable en milieu basique et ne présente pas de caractère réducteur.
|
Utilisation comme groupe protecteur
On utilise souvent un diol comme l'éthane-1,2-diol car avec ces composés, on obtient des acétals cycliques. La réaction est alors thermodynamiquement moins défavorable grâce à l'effet entropique (deux molécules conduisent à deux molécules).
La séquence réactionnelle suivante illustre l'utilisation d'un groupe protecteur pour le groupe carbonyle dans une synthèse magnésienne d'alcool :
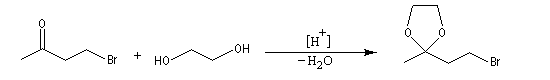
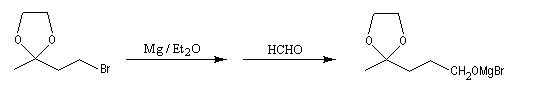
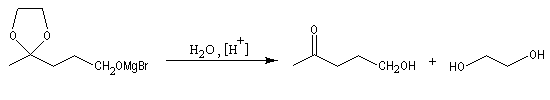
Un mode opératoire de réaction de blocage d'un carbonyle par l'éthane-1,2-diol est donné à la référence [13].
Les diols peuvent être protégés par formation d'un acétal avec l'acétone qui est peu coûteuse. Ce mode de protection est utilisé notamment dans la chimie des sucres.
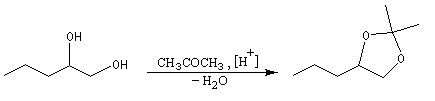
En série cyclique, les groupes OH vicinaux doivent être en position cis, l'un par rapport à l'autre. Dans l'exemple ci-dessous, la formation du cétal est régiosélective.
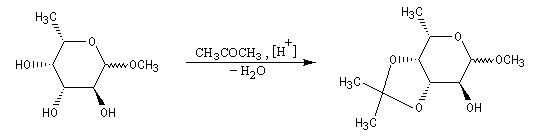
La réaction ci-dessous est une étape de la synthèse du cholestérol (Woodward 1952 [36].)
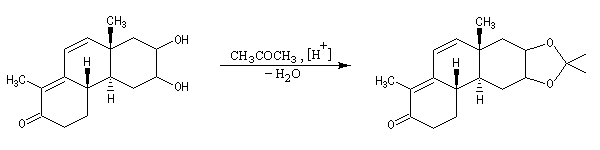
Une méthode pratique de protection des alcools consiste à les faire réagir avec un éther d'énol comme le dihydropyrane (DHP) ce qui conduit à la formation d'un cétal.
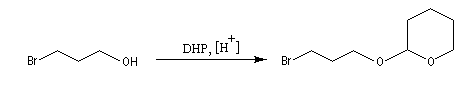
L'acétalisation peut être thermodynamiquement favorisée lorsqu'elle est intramoléculaire car l'effet entropique est très favorable. Certains acétals existent à l'état naturel. La frontaline est une phéromone d'agrégation de coléoptères appartenant à la famille des scolitidae. Parmi ces insectes, le scarabée Dendroctonus frontalis Zimmermann (Southern Pine Beetle) est l'insecte le plus destructeur de forêts de pins dans le sud des USA.
Bilan
Une réaction d'acylation consiste formellement à substituer l’atome d’hydrogène du groupe -OH par un groupement acyle R-CO-. On peut y parvenir en effectuant la réaction entre l'alcool et l'acide carboxylique ou l'un de ses dérivés : halogénure d'acyle, anhydride ou ester.
Avec les deux premiers, la réaction est à la fois totale et rapide. Avec l'acide, elle conduit à un équilibre qu'on peut déplacer dans le sens de la formation de l'ester. Elle nécessite l'utilisation d'un catalyseur. La réaction entre un alcool et un ester est appelée transestérification. Les hydroxyacides conduisent aux lactones par estérification intramoléculaire.
Acylation par un chlorure d'acyle ou un anhydride
On effectue la réaction entre l'alcool et le chlorure d'acyle ou l'anhydride en présence d'une amine tertiaire comme la pyridine ou la diméthylaminopyridine DMAP (catalyseur de Steglich).
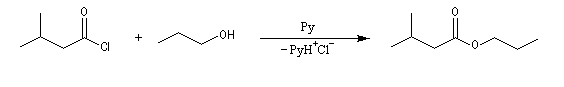
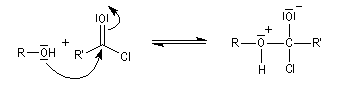
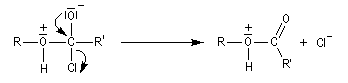
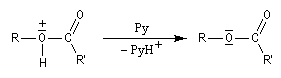
Dans cette réaction, la pyridine est plus qu'une simple base. Elle joue aussi un rôle cinétique. C'est un catalyseur nucléophile.
Plus précisément, la réaction entre le chlorure d'acyle et la pyridine fournit un ion acylaminium intermédiaire I.
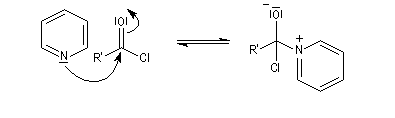
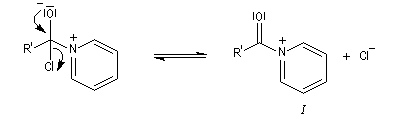
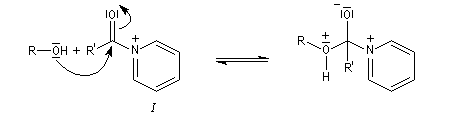
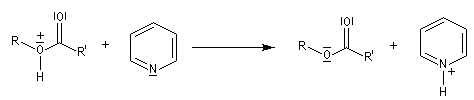
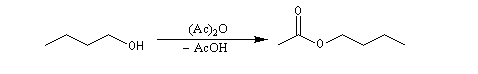
Utilisation des esters comme groupe protecteurs
Les esters peuvent être utilisés comme groupes protecteurs de la fonction alcool. Il existe de nombreuses méthodes. L'utilisation d'un anhydride fluoré est efficace.
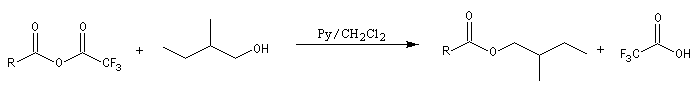
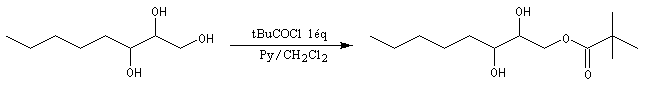
La déprotection s'effectue par hydrolyse acide ou basique et dans certains cas par hydrogénolyse
Acylation par un acide carboxylique
La réaction entre un acide carboxylique et un alcool est appelée estérification de Fischer. On obtient un ester et de l'eau.
La réaction entre l'acide butanoïque et le méthanol s'écrit :
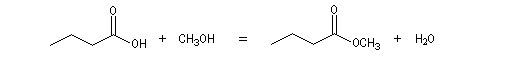
On peut étudier la réaction d'estérification d'un double point de vue :
La réaction étant quasiment athermique, on ne peut espérer déplacer la position de l'état d'équilibre en élevant la température. Pour favoriser la formation d'ester, on peut utiliser un excès du réactif le moins cher ou éliminer l'un des produits au fur et à mesure de sa formation. Il y a deux possibilités :
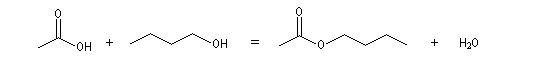
|
|
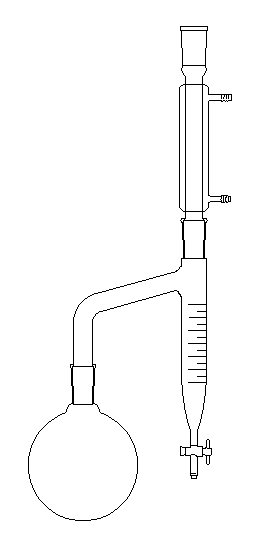
Le dessin de gauche et la photographie ci-dessous représentent un montage permettant la préparation d'un ester en utilisant un décanteur de type Dean-Stark.
Dans le ballon, on réalise un mélange d'acide, d'alcool et de toluène (on utilise aussi le benzène mais l'usage de ce dernier répond à une réglementation précise du fait de sa toxicité). |
L'eau et le toluène ne sont pas miscibles à l'état liquide et ils forment un hétéroazéotrope. Le mélange de vapeur d'eau et de toluène monte dans le réfrigérant ascendant. Lorsque la température diminue, les vapeurs se liquéfient pour donner deux liquides non miscibles. L'eau, plus dense, tombe au fond du décanteur. Elle peut être extraite du milieu réactionnel au fur et à mesure de sa formation.
Un mode opératoire possible pour la préparation de l'éthanoate de butyle est donné ci-dessous :
On introduit dans le ballon 0,25 mol d'acide acétique (éthanoïque d = 1,05) et 0,25 mol de butan-1-ol (d = 0,81). On ajoute 30 mL de toluène et environ 0,15 g d'acide paratoluènesulfonique APTS ainsi que quelques grains de pierre ponce.
Le mélange est chauffé avec un chauffe-ballon tant que l'eau est entraînée.
Avec un appareil de Dean-Stark gradué on peut tracer la courbe donnant V en fonction du temps (remarque : si l'on trace V en fonction de 1/t , on obtient pratiquement une droite).
En série cyclique, la vitesse d'acétylation d'alcools tels que le tertiobutylcyclohexanol dépend de l'orientation axiale ou équatoriale du groupe hydroxyle.
Mécanisme de l'estérification des alcools primaires et secondaires AAC2
Le marquage isotopique (18O) de l'oxygène de l'alcool, suivi de l'analyse par spectrométrie de masse des produits, montre que cet atome se retrouve dans l'ester.
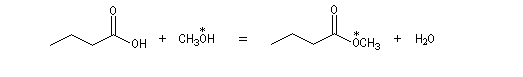
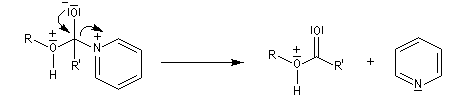
Le mécanisme suivant s'applique aux alcools primaires et secondaires. Il s'agit d'un mécanisme par stades comportant une addition suivie d'une fragmentation. On distingue les différentes étapes suivantes qui sont renversables :
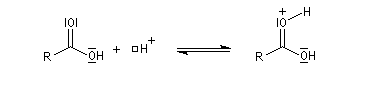
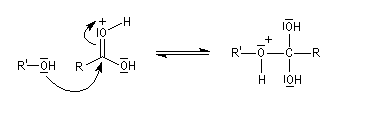
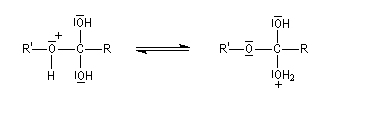
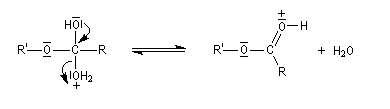
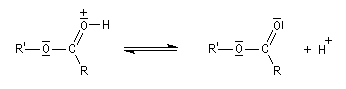
L'étape cinétiquement déterminante est la formation de l'intermédiaire tétraédrique. Il s'agit d'une réaction bimoléculaire. Comme la liaison qui se romp lors de l'étape de fragmentation provient de l'acide, Ingold a appelé AAc2 ce mécanisme.
Mécanisme de l'estérification des alcools tertiaires AAL1
Les alcools tertiaires comme le 2-méthylpropan-2-ol peuvent également être estérifiés en utilisant comme agent acylant un anhydride d'acide ou un chlorure d'acyle et un catalyseur tel que ZnCl2.
L'exemple suivant concerne la préparation de l'éthanoate de 2-méthylpropyle dont on trouvera un mode opératoire à la référence [24].
Le mécanisme est un cas particulier de mécanisme SN1 qui est noté AAL1 car la liaison qui se rompt dans l'étape
cinétiquement déterminante est celle de l'alcool, cette étape étant monomoléculaire.
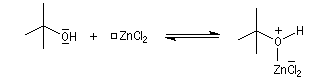
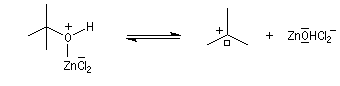
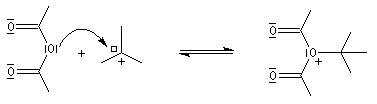
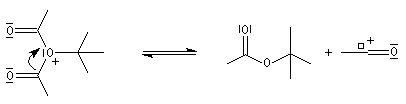
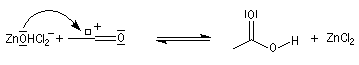
La réaction entre un alcool et un ester est appelée transestérification. Elle est étudiée dans le chapitre relatif aux esters.
Méthylation par le diazométhane
Les alcools peuvent être méthylés par le diazométhane en présence d'un acide de Lewis comme BF3. Exemple [27]. Cette réaction nécessite des précautions particulières car le diazométhane est un agent méthylant très toxique.![]()
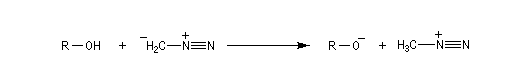
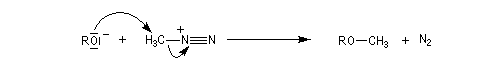
Une réaction du même type permet la méthylation des ions carboxylate.
Estérification par l'acide nitrique
La nitroglycérine est le représentant le plus connu des nitrates d'alkyles. Ces composés sont tous très instables et donc extrêmement dangereux à manipuler.
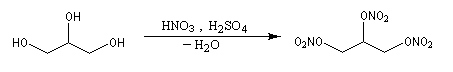
Comme pour la plupart des explosifs, une petite quantité de nitroglycérine libère un très grand volume de gaz.
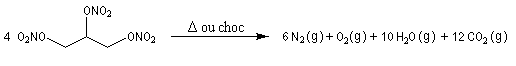
|
|
La nitroglycérine fut préparée pour la première fois en 1846 par le chimiste italien A. Sobrero. Ce composé est particulièrement instable et peut exploser sous l'action d'un choc. Le suédois A. Nobel découvrit en 1866 qu'on pouvait stabiliser la nitroglycérine en la mélangeant à un sable siliceux d'origine naturelle : le Kieselguhr. La nitroglycérine est utilisée en médecine comme vasodilatateur sous le nom plus rassurant de trinitrine. Les intérêts de la fortune accumulée par A. Nobel servent à distribuer les prix Nobel qui sont attribués depuis 1900 par l'Académie royale de Suède. |
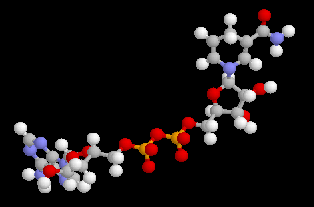
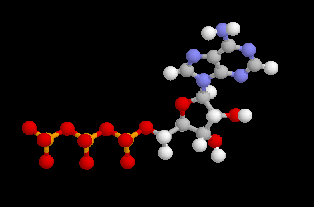
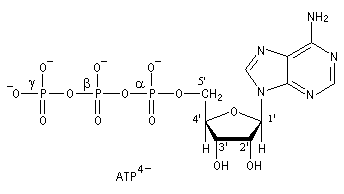
Estérification par l'adénosine triphosphate ATP4-
L'adénosine triphosphate (ATP) est un coenzyme nucléotidique. Dans les conditions de pH rencontrées en milieu biologique, l'ATP est essentiellement sous forme ATP4- : pKa (HATP3-/ATP4-) = 6,5.
La phosphorylation du glucose par l'ATP en glucose-6-phosphate est une importante réaction biochimique intervenant dans la glycolyse (coupure du glucose en pyruvate). Elle est catalysée par une enzyme spécifique, l'hexokinase.
|
|
On peut distinguer trois parties dans l'ATP : l'association de l'adénine et du ribose constitue le nucléoside adénosine. Il y a ensuite une succession de trois groupes phosphate associés au groupe 5'-OH de l'adénosine par une liaison phosphoester. Les groupes phosphate sont liés entre-eux par des liaisons phosphoanhydrides. L'ATP constitue la réserve principale d'énergie chimique de la cellule vivante. L'hydrolyse des liaisons phosphoanhydrides est fortement exergonique : Pour l'hydrolyse de l'ATP en ADP : DrG0' = -34,5 kJ.mol-1 Notons que dans la cellule, le coenzyme est complexé avec des ions Mg2+ lié aux phosphates a et b . |
Coupure C-O
Généralités
Le groupe hydroxyle d'un alcool est un mauvais nucléofuge ce qui est à mettre en relation avec son caractère de base forte. La protonation du groupe hydroxyle accroît considérablement les possibilités de rupture de la liaison carbone-oxygène car le nucléofuge est maintenant une petite molécule stable : l'eau.
Formation de carbocations
La rupture de la liaison carbone-oxygène peut conduire à un carbocation. C'est le cas avec les alcools tertiaires. Certains peuvent être suffisamment stables pour pouvoir être observés. Il est ainsi très facile d'obtenir le carbocation triphénylméthyle à partir du triphénylméthanol par ajout d'acide sulfurique.
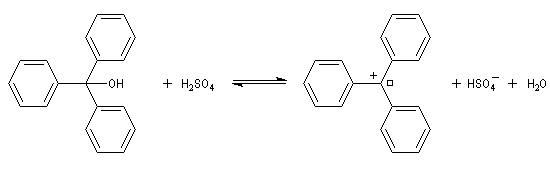
Dans le cas précédent on peut identifier le carbocation grâce à la couleur rouge que prend le milieu réactionnel.
|
|
A la température ordinaire, le triphénylméthanol est un solide de couleur blanche (TF = 136 °C). Dans le bécher j'ai mis une petite quantité de triphénylméthanol solide. L'ajout de quelques gouttes d'acide sulfurique concentré provoque l'apparition d'une couleur rouge intense. Cette expérience, réalisée en 1901 indépendamment par Norris et Kehrmann, fut la première mise en évidence de l'intervention de carbocations en chimie organique. L'acide sulfurique protone l'alcool et permet le départ du nucléofuge H2O. L'équilibre est déplacé vers la droite en raison de la stabilité du cation triphénylméthyle (carbocation trityle) et du caractère déshydratant de l'acide sulfurique concentré qui capte l'eau formée. |
Le carbocation triphényleméthyle peut aussi être obtenu en effectuant la réaction de Friedel et Crafts entre le benzène et le tétrachlorométhane en présence d'un acide de Lewis suffisamment puissant comme AlCl3.
Structure du cation trityle : la structure du carbocation triphényleméthyle (carbocation trityle) dans un composé solide, a été déterminée par diffraction des rayons X dans les années 60 (A.H. Gomes et C.H. Mac Gillavry). Les liaisons partant de l'atome de carbone central sont dans un même plan. Du fait de la répulsion des atomes d'hydrogène situés en ortho, les cycles adoptent une conformation non plane et font des angles de 54° par rapport au plan de ces liaisons. Globalement, le cation a la forme d'une hélice qui rappelle celle du
radical triphénylméthyle. L'existence d'une lacune électronique portée par l'atome de carbone central permet une délocalisation des électrons sur un système de grande dimension. Cette délocalisation importante est à l'origine de la stabilité relativement grande du carbocation.Le cation triphénylméthyle forme un système conjugué de grandes dimensions. Ce système absorbe la lumière dans le domaine visible d'ou la couleur observée qui est approximativement complémentaire de celle qui est absorbée.
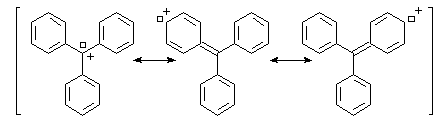
On retrouve des systèmes conjugués comparables dans de nombreuses matières colorantes comme le vert malachite.
Mis à part le cas particulier étudié, les carbocations sont des espèces très réactives, qui n'apparaissent généralement que comme intermédiaire dans des réactions de substitution ou d'élimination.
Halogénation par les hydracides halogénés
Alcools primaires
Avec HBr et HI, on peut synthétiser des dérivés halogénés à partir des alcools primaires. H+ protone l'alcool et I- ou Br- sont suffisamment nucléophiles pour déplacer l'eau par substitution nucléophile bimoléculaire. On peut aussi utiliser KI et KBr en milieu H2SO4 concentré. Cela revient moins cher que d'utiliser des acides halohydriques.
![]()
En milieu acide le groupe OH est protoné ce qui améliore l'aptitude nucléofuge du groupe sortant.
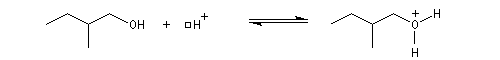
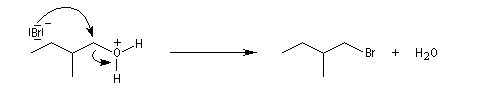
La réaction entre HCl concentré et un alcool primaire ne conduit pas au dérivé halogéné même à chaud car Cl- n'est pas assez nucléophile. En présence d'un catalyseur comme ZnCl2, le butan-1-ol fournit le chlorobutane après un reflux de plusieurs heures.
![]()
L'alcool réagit avec ZnCl2 qui est un acide de Lewis.
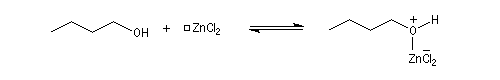
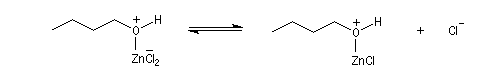
Le groupe -OH est remplacé par -O+HZnCl qui est un bien meilleur nucléofuge. Le mécanisme est du type SN2.
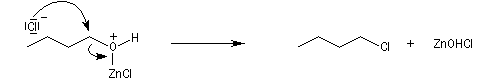
Le catalyseur est régénéré.
![]()
Un inconvénient de ce type de réactions est qu'elles s'accompagnent souvent de transpositions de type Wagner-Meerwein. Elles sont assez peu utilisées pour préparer les halogénures d'alkyles.
![]()
![]()
![]()
![]()
Une autre réaction secondaire est l'élimination d'un proton à partir du carbocation pour former un composé éthylénique.
Alcools tertiaires
Un alcool tertiaire comme le 2-méthylpropan-2-ol (tertiobutanol) est transformé en chlorure par simple agitation avec l'acide chlorhydrique concentré à froid. Le mécanisme est une substitution nucléophile monomoléculaire SN1 avec formation d'un carbocation intermédiaire.
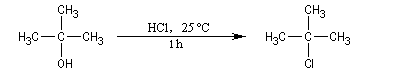
En milieu acide l'alcool est protoné.
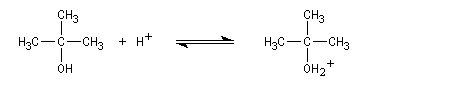
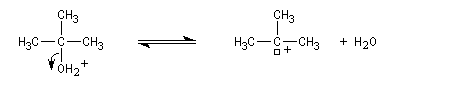
Le carbocation réagit rapidement avec le nucléophile.
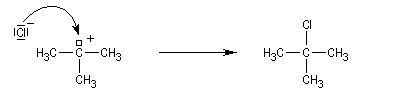
La réaction s'accompagne de la formation d'une petite quantité de composés éthyléniques car le carbocation formé peut aussi évoluer par élimination E1.
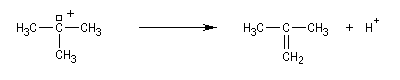
Alcools secondaires
Les alcools secondaires ont une réactivité intermédiaire entre celle des alcools primaires et celle des alcools tertiaires. Ils sont assez plus rapidement transformés en halogénure à chaud par HI, HBr ou le mélange HCl, ZnCl2 que les primaires.
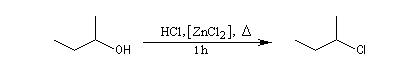
Le mécanisme est de type SN1. Le passage par des carbocations intermédiaires, entraîne des transpositions de type Wagner-Meerwein.
Test de Lucas des alcools
Le rôle catalytique de ZnCl2 dans l'halogénation par les ions chlorure est à la base d'un test des classes d'alcools mis au point par le chimiste américain H. J. Lucas. Le réactif de Lucas est une solution de ZnCl2 dans l'acide chlorhydrique concentré. On effectue un mélange de l'alcool à tester et du réactif. Le test repose sur la différence de réactivité des alcools des différentes classes vis à vis de la coupure C-O. Un test positif se traduit par l'apparition de deux phases car l'halogénure formé est peu miscible avec le mélange des réactifs.
|
Classe de l'alcool |
Primaire |
Secondaire |
Tertiaire |
|
Vitesse |
très lent à chaud |
rapide à chaud |
rapide à froid |
La vitesse de la réaction est d'autant plus grande que le substrat peut mieux stabiliser la charge positive qui se développe sur l'atome de carbone. Les alcools tertiaires qui donnent facilement des carbocations réagissent donc les plus rapidement.
Halogénation par des réactifs inorganiques
Réactifs halogénants
Les alcools peuvent être convertis en dérivés halogénés grâce à une assez grande variété de réactifs halogénants :
Un réactif couramment employé est le chlorure de thionyle SOCl2.
|
Le chlorure de thionyle peut être préparé par réaction entre SO2 et PCl5. On obtient SOCl2et POCl3 qui sont ensuite séparés par distillation fractionnée. L'arrangement des doublets autour du soufre est tétraédrique (structure AX3E au sens de la théorie VSEPR). La molécule a la forme d'une pyramide irrégulière. Ce composé est hydrolysé de façon violente par l'eau en donnant HCl et SO2. C'est un réactif qu'il faut manipuler avec précautions. |
La réaction est souvent conduite en présence d'une amine tertiaire comme la pyridine pour capter HCl formé. Le dioxyde de soufre SO2 est un gaz dans les conditions de l'expérience. Il faut prévoir un piège à gaz acide. La méthode est utilisable avec les alcools primaires et secondaires.
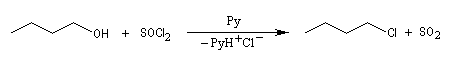
Les halogénures de phosphore sont des agents halogénants très utilisés.
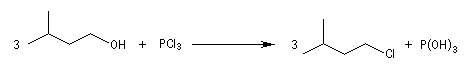
| Le trichlorure de phosphore PCl3 peut être préparé par union directe entre le phosphore (rouge) et le dichlore. C'est un composé très hygroscopique. Son hydrolyse par l'air humide produit HCl. Elle s'accompagne d'un brouillard (dû à l'existence d'un azéotrope eau-HCl). On dit par abus de langage qu'il fume à l'air. La molécule est pyramidale (voir la méthode VSEPR). |
Avec PBr3 et PI3 des réactions analogues permettent d'accéder aux dérivés bromés et iodés. Dans le dernier cas on réalise le mélange P (rouge) et I2 qui forme PI3 in situ, ce composé étant instable.
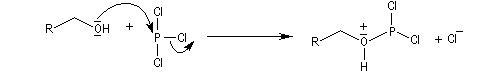
L'ion chlorure déplace le nucléofuge grâce à une substitution nucléophile bimoléculaire.
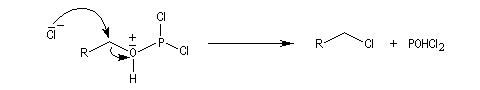
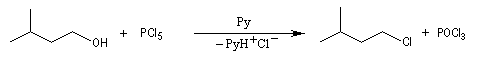
On obtient comme sous-produit POCl3 qu'il faut ensuite séparer du milieu réactionnel par distillation. Ce dernier peut réagir avec l'excès d'alcool.
|
Le pentachlorure de phosphore peut être obtenu par réaction entre PCl3 et Cl2. A l'état vapeur, il est constitué de molécules de formule PCl5. La molécule à la forme d'une bipyramide trigonale (voir la méthode VSEPR). A l'état solide, le pentachlorure de phosphore est constitué de cations PCl4+ et d'anions PCl6-. On continue à écrire PCl5 pour simplifier. Il est particulièrement hygroscopique et violemment hydrolysé par l'eau. |
Cette méthode ne s'applique pas aux alcools possédant des substituants en b. L'inconvénient est le faible pourcentage d'utilisation de l'élément chlore.
Sur un substrat chiral, on observe l'inversion de configuration du centre chiral. L'exemple suivant, utilisant la projection de Fischer, concerne une réaction extraite du travail classique de Walden sur la chloration des acides maliques.
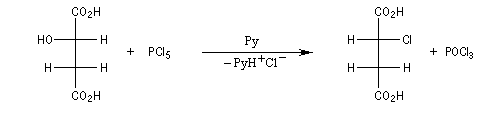
Aspect stéréochimique
Lorsqu'elle est effectuée en présence de pyridine, la réaction entre le (2S)-octan-2-ol et SOCl2 fournit le (2R)-2-chlorooctane. La réaction est une substitution nucléophile bimoléculaire SN2. On observe l'inversion de Walden qui s'accompagne ici d'une modification de la configuration absolue du centre stéréogène.
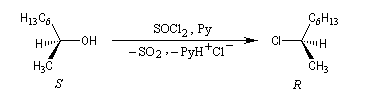
La réaction entre le (2S)-octan-2-ol et SOCl2 peut être effectuée en l'absence de pyridine. En présence d'éther comme solvant, on obtient le (2S)-2-chlorooctane. Dans ce cas la configuration est conservée. Cette rétention de configuration se traduit ici par l'invariance de la configuration absolue du centre stéréogène.
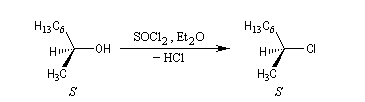
On interprète ce résultat par une substitution nucléophile interne SNi au sein de la paire d'ions qui résulte de la décomposition du chlorosulfite intermédiaire. Avec un substrat comme l'octan-2-ol, la présence d'éther est indispensable car ce solvant stabilise la paire d'ions.
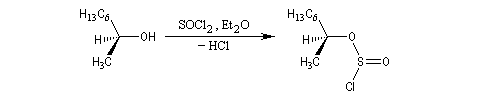
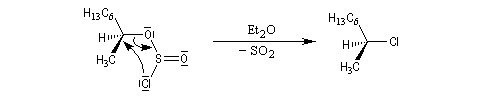
Avec le phényléthanol qui forme un carbocation benzylique assez stable, on peut observer le mécanisme SNi même en l'absence d'éther.
Bilan et conditions expérimentales
Une solution pour améliorer le caractère nucléofuge du groupe hydroxyle est de le remplacer par un autre groupe ! Le chlorure de paratoluènesulfonyle (TsCl) est un dérivé de l'acide paratoluènesulfonique (APTS).
|
|
L'acide paratoluènesulfonique (APTS) peut être obtenu par réaction de sulfonation entre le toluène et l'acide sulfurique. A la température ordinaire, il se présente sous la forme d'un solide. Il s'agit d'un acide fort : pK (TsOH/TsO-) = -7. L'APTS est souvent utilisé comme catalyseur acide car il est soluble en milieu organique. La base conjuguée correspondante n'est pas nucléophile ce qui limite les réactions secondaires. De plus, contrairement à l'acide sulfurique il n'est pas du tout oxydant.
La base conjuguée de l'APTS, l'ion paratoluènesulfonate ou tosylate est un excellent nucléofuge (base très faible). |
On prépare le chlorure de paratoluènesulfonyle ou chlorure de tosyle, grâce à la réaction suivante :
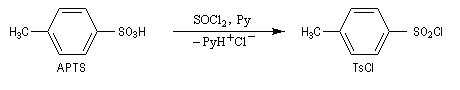
La réaction entre TsCl et un alcool conduit au paratoluène sulfonate ROTs souvent appelé tosylate.
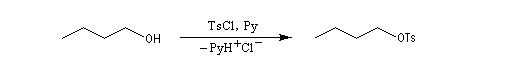
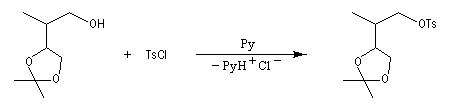
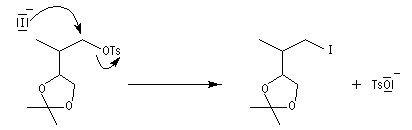
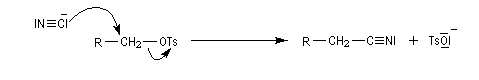
Notons que cette réaction ne serait pas possible en milieu acide car l'ion cyanure serait protoné pour donner HCN. La méthode est surtout valable pour les alcools primaires et secondaires.
En milieu biologique les composés contenant des groupes hydroxyles peuvent être activés par transformation en groupes phosphates. Par exemple par formation d'uridinediphosphoglucose (UDPG).
Elimination : passage aux composés éthyléniques
Bilan, conditions expérimentales
Un moyen très simple pour préparer le cyclohexène consiste à chauffer le cyclohexanol avec de l'acide sulfurique ou de l'acide phosphorique concentrés. La réaction s'écrit :
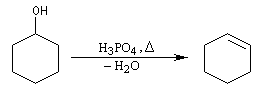
Cette réaction est générale. Les alcools conduisent aux composés éthyléniques par élimination d'eau. On peut regarder cette réaction comme l'inverse de l'hydratation de ces composés.
L'élimination peut être effectuée en présence d'un acide fort comme H2SO4 ou mieux H3PO4 qui ne présente pas l'inconvénient par rapport au précédent d'oxyder le substrat [2]. En milieu acide, l'alcool est protoné ce qui permet le départ d'eau bien meilleur nucléofuge que OH. On peut également utiliser des acide de Lewis tels que ZnCl2, BF3, I2 comme catalyseur de déshydratation. Un mode opératoire pour la préparation du cyclohexène se trouve dans [6].
Influence de la classe d'alcool
Des conditions typiques d'élimination à partir d'alcools de différentes classes sont les suivantes :
|
Classe d’alcool |
Réactif |
Température (°C) |
|
Primaire |
H2SO4 (98 %) |
180 |
|
Secondaire |
H2SO4 (50 %) |
140 |
|
Tertiaire |
H2SO4 (20 %) |
80 |
La réaction est d'autant plus facile que la classe de l'alcool est plus élevée. Avec un alcool tertiaire un léger chauffage en présence d'acide dilué suffit à provoquer l'élimination.
Compétition entre élimination et substitution
Lorsqu'on chauffe de l'éthanol en présence d'acide sulfurique à 140 °C, on obtient essentiellement de l'éthoxyéthane. A 180 °C le produit majoritaire est l'éthène.
|
T (°C) |
Réactif |
Produit |
Type de réaction |
|
140 |
H2SO4 (98 %) |
Ethoxyéthane |
Intermoléculaire |
|
180 |
H2SO4 (98 %) |
Ethène |
Intramoléculaire |
Les éthers simples symétriques peuvent être synthétisés par élimination d'eau entre deux molécules d'alcool.
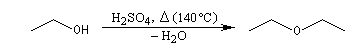
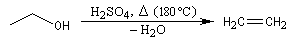
Le problème ne se pose pas pour les alcools tertiaires qui ne réagissent pas par substitution. Dans les autres cas, l'élimination est favorisée par rapport à la substitution lorsque la température augmente. On peut le comprendre en remarquant que dans le premier cas il y a deux liaisons à rompre alors que dans le deuxième cas, il n'y en a qu'une. Il faut donc fournir davantage d'énergie pour promouvoir l'élimination par rapport à la substitution.
Mécanismes
La déshydratation d'un alcool peut être regardée comme la réaction inverse de l'hydratation d'un alcène acido-catalysée. Avec les alcools tertiaires et secondaires, il y a formation d'un carbocation. Il s'agit d'un mécanisme de type E1. La protonation de la fonction alcool permet de transformer le mauvais groupe libéralble OH en
un meilleur nucléofuge : l'eau.
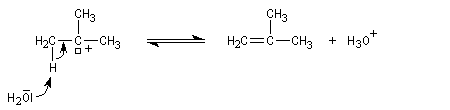
Régiosélectivité
On s'intéresse à la déshydratation catalysée par un acide, du 2-méthylbutan-2-ol.
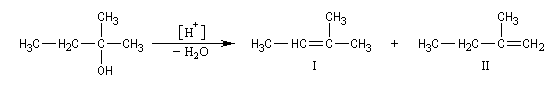
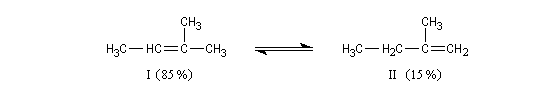
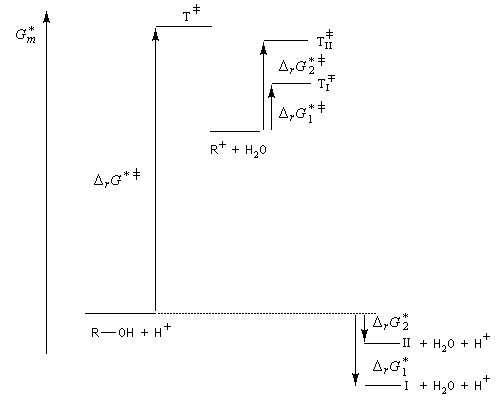
La situation est donc différente de celle qu'on observe lors des déshydrohalogénations E1 des dérivés halogénés qui sont sous contrôle cinétique bien que la règle de Zaytzev soit suivie.
Stéréosélectivité
La déshydratation du butan-2-ol dans H2SO4 à 60 % à 25 °C fournit plusieurs composés avec les pourcentages suivants.
|
But-1-ène |
(Z)-But-2-ène |
(E)-But-2-ène |
|
3 % |
23 % |
74 % |
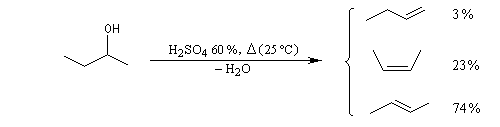
La stéréosélectivité s'explique également par un contrôle thermodynamique de la réaction. La déshydratation du butan-2-ol fournit de façon majoritaire l'un des deux alcènes diastéréo-isomères possibles. L'alcène de configuration E, plus stable que celui de configuration Z est obtenu majoritairement.
En revanche le (2R)-butan-2-ol et le (2S)-butan-2-ol qui sont énantiomères, fournissent le même alcène. La réaction n'est pas stéréospécifique.
Le passage par des carbocations explique l'existence de transpositions fréquentes dans ce type de réaction.
Recherche de structure
Dans certains cas, les réactions d'élimination peuvent être mises à profit pour préciser la structure des alcools. Dans l'exemple ci-dessous, la position de la double liaison éthylénique dans les produits de déshydratation des alcools A et B peut être déterminée grâce à la réaction d'ozonolyse. Il est alors facile de préciser la structure de l'alcool de départ.
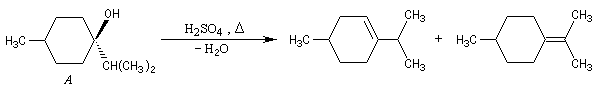
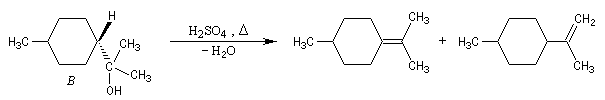
Si l'on obtient du méthanal dans les produits issus de l'ozonolyse, l'alcool de départ est B.
Transpositions
Transposition de carbocations
On peut aussi assister à une réaction de transposition du carbocation. Dans l'exemple ci-dessous, celle-ci s'accompagne d'un agrandissement de cycle.
![]()
![]()
![]()
![]()
Il s'agit d'un cas particulier de transposition de Wagner-Meerwein.
Transposition pinacolique
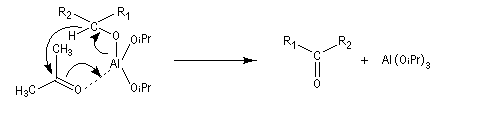
Lorsqu'on traite en milieu acide le 2,3-diméthylbutane-2,3-diol, appelé communément pinacol, on observe une réaction de transposition conduisant à la 3,3-diméthylbutan-2-one. Cette réaction, qui constitue un des premiers exemples de transposition mis en évidence en chimie organique, s'appelle transposition pinacolique [20]. Notons que le pinacol peut être facilement obtenu par réduction duplicative de la propanone.
Le mécanisme de la transposition est le suivant :
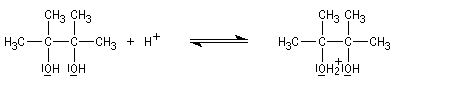
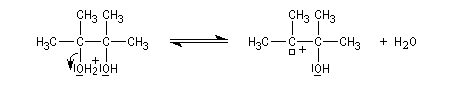
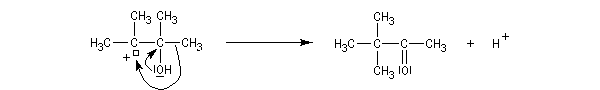
Notons qu'au cours de cette étape, il y a passage d'un cation tertiaire à un cation secondaire plus stable car il est substitué par un atome d'oxygène donneur par effet mésomère.
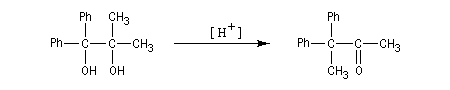
Dans le deuxième exemple, le carbocation se forme au même endroit mais cette fois, le groupe phényle migre de préférence au méthyle.
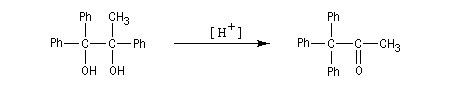
Notons que les groupes aromatiques substitués par des substituants donneurs (méthoxy, alkyle) ont une aptitude migratoire supérieure à ceux qui sont liés à des substituants attracteurs (halogènes, nitro). En effet, les premiers stabilisent davantage une charge positive que les seconds.
La transposition pinacolique peut également avoir lieu de façon intramoléculaire. La réaction suivante est l'une des étapes de la synthèse du longifolène par E. J.Corey.
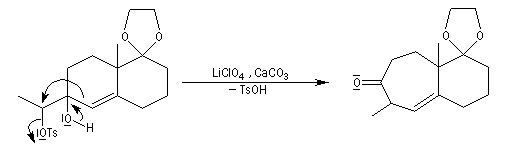
Bibliographie
Ouvrages et articles expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[13] D. R. Paulson, Journal of Chemical Education, 50, 1973.
[14] Dess, D. B. ; Martin, J. C. J. Org. Chem. 1983, 48, 4155-4156.
[15] Dess, D. B. ; Martin, J. C. J. Am. Chem. Soc. 1991, 1, 7277-7287.
[16] Corey, E. J. ; Mitra, R. B. ; Vatakencherry, P. A. ; J. Am. Chem. Soc. 86, 478 (1964).
[28] Corey E. J., C. U. Kim C. U., J. Am. Chem. Soc. 94, 7586 (1972).
[29] Mancuso, A. J. Swern, D. Synthesis, 1981, 165-185.
[35] Parikh, J. R., Doering, W. Von E, J. Am. Chem. Soc. 1967, 5505-5507.
[36] R. B. Woodward, F. Sondheimer, D. Taub, K. Heusler and W. M. McLamore J. Amer. Chem. Soc. 74, 4223 (1952.)
[37] R. Criegee, Ber. 64, 260 (1931.)
[17] Dess-Martin reagent
[18] Swern Oxidation
[19] Oxydation d'Oppenauer du cholestérol
[20] Pinacolone
[21] Moffatt Oxidation ; 4-tbutylcyclohexanone
[23] Aldehydes from primary alcohols : heptanal by J. C. Collins and W. W. Hesse
[25] Dess-Martin periodinane by R. K. Boeckman, P. Shao and J. J. Mullins
[26] Syntheses of (Diacetoxyiodo)arenes or Iodylarenes from Iodoarenes, with Sodium Periodate as the Oxidant by Pawel Kazmierczak, Lech Skulski, and Lukasz Kraszkiewicz
[27] Cholestanyl methyl ether by M. Neeman and William S. Johnson.
[30] Oxidation of alcohols by methylsulfide, N-chlorosuccinimide, triéthylamine by E. J. Corey, C. U. Kim and P. F. Misco.
[31] o-iodoxybenzoic acid IBX ; SIBX
[32] Cours de G. Myers, University of Harvard Chapitre très complet sur les réactions d'oxydation des alcools et d'autres fonctions. Nombreuses références bibliographiques.
[33] Oxydations en chimie organique
A. Streitwieser Jr, C.H. Heathcock - Introduction to Organic Chemistry (Macmillan Publishing Co).
J. March - Advanced organic chemistry (Wiley Interscience).
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, (Plenum Press 1990).
N. T. Ahn - Introduction à la chimie moléculaire, (Ellipses 1994).
P. Caubère - Les dérivés du soufre en chimie organique, (Masson).
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale (Hermann 1982).
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique (Ellipses - 1993).
P. Atkins - General Chemistry (Scientific American Books W.H Freeman and Co).
J. Koolman, K.H Röhm - Atlas de biochimie (Médecine & Sciences ; Flammarion).
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
mai 2014