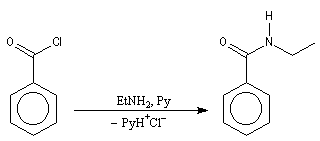
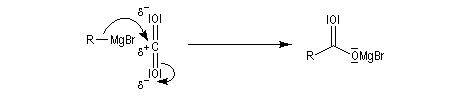
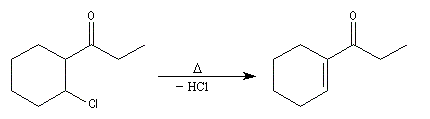
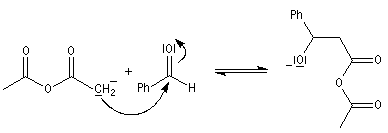
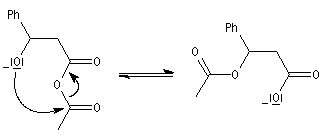
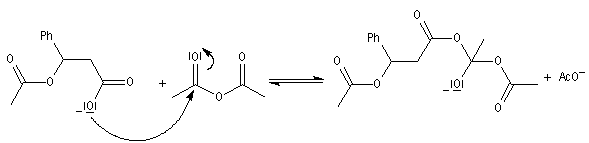
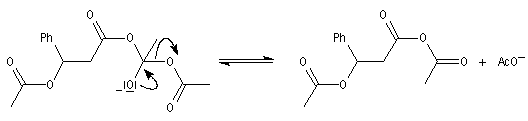
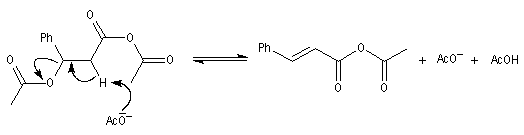
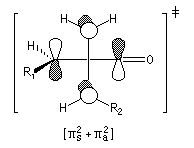
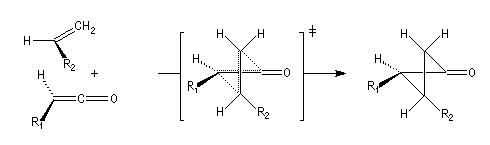
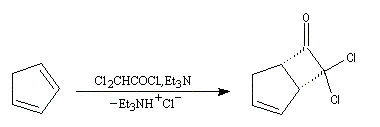
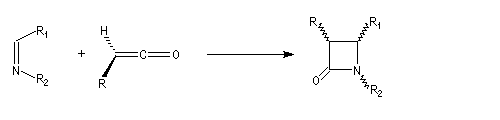
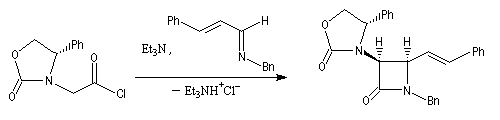
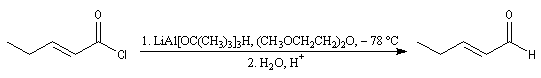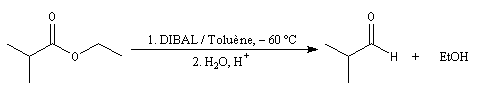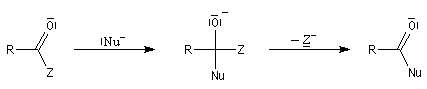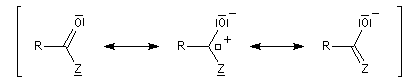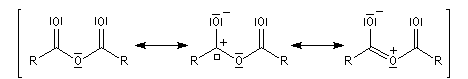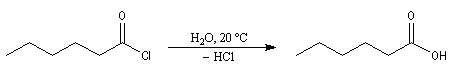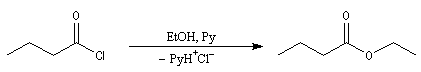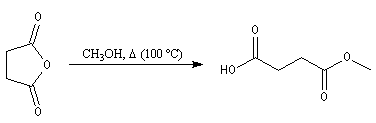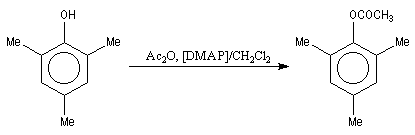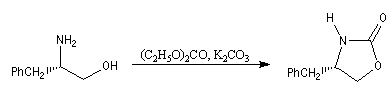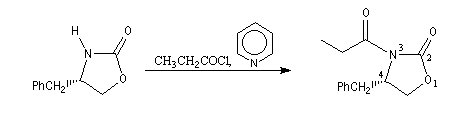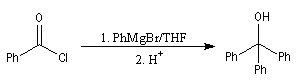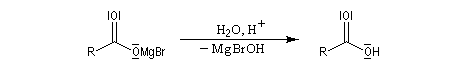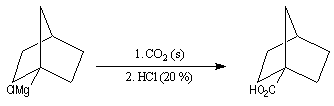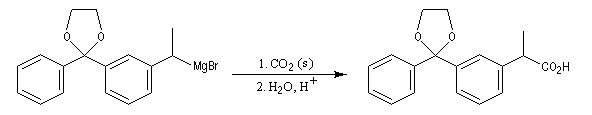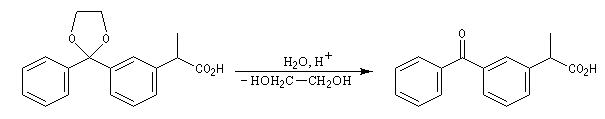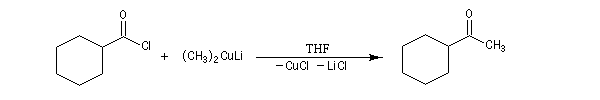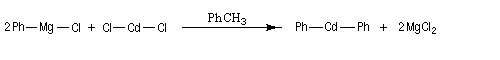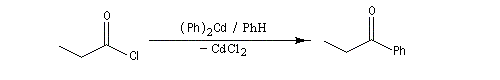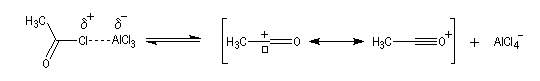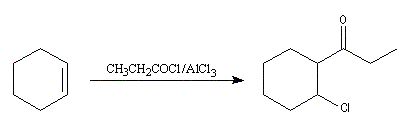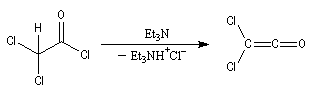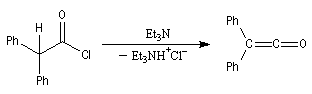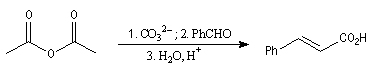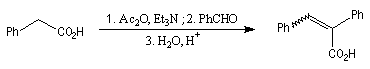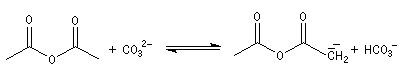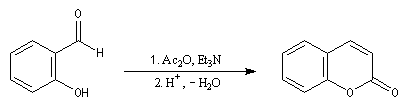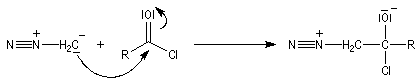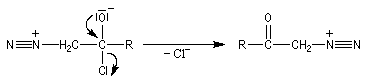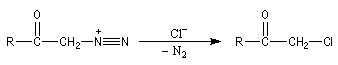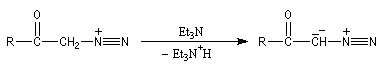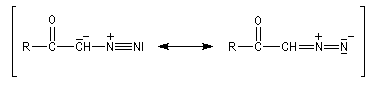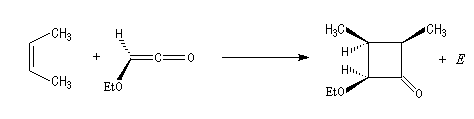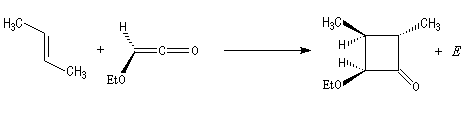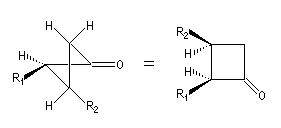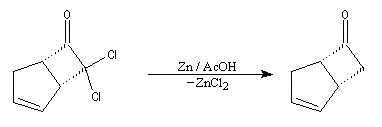|
Formule |
|
|
|
|
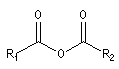
Composé |
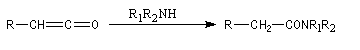
cétène |
anhydride |
halogénure d'acyle |
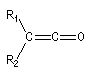
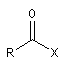
|
|
|
|
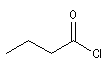
chlorure de butanoyle |
bromure d'hexa-4-énoyle |

|
|
|
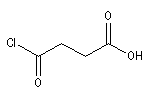
acide 3-chloroformylpropanoïque |
Anhydrides d'acides anhydride éthanoïque anhydride éthanoïque et propanoïque anhydride butane-1,4-dicarboxylique anhydride pentane-1,5-dicarboxylique Propriétés physiques Anhydrides Composé anhydride éthanoïque anhydride propanoïque TE (° C) 140 168 Halogénures d'acyles Composé chlorure d'éthanoyle chlorure de propanoyle TE (° C) 52 80 Le dichlorure d'acyle de l'acide carbonique ou phosgène est un composé gazeux à la température ordinaire qui fut
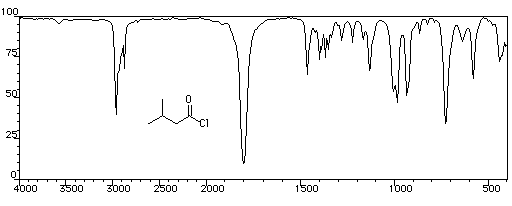
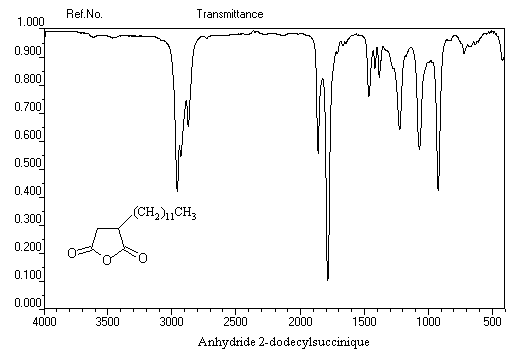
utilisé comme gaz de combat pendant la première guerre mondiale. Spectroscopie Spectroscopie infrarouge Groupe fonctionnel C = O C-Cl s (cm-1) 1785 - 1815 875 La conjugaison entre le chlore et le carbonyle entraîne pour ce dernier un effet hypsochrome par rapport au groupement carbonyle d'une cétone. La vibration d'élongation
de C-Cl, présente un harmonique vers 1745 cm-1 Il est intéressant de comparer les absorptions du groupement carbonyle des cétones, des esters et des anhydrides. Groupe fonctionnel cétones esters anhydrides s (cm-1) 1705 - 1725 1735 - 1750 1800 - 1850
On distingue les anhydrides symétriques et les anhydrides mixtes. Les premiers sont nommés en remplaçant le mot acide par anhydride.
Les anhydrides mixtes sont nommés en faisant suivre le mot anhydride des noms des réactifs parents, classés dans l'ordre alphabétique.
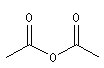
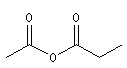
Les composés cycliques sont nommés de préférence comme des anhydrides et non comme des hétérocycles
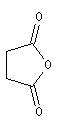
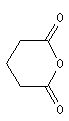
Ce sont des composés polaires dont la température d'ébullition est légèrement supérieure à celle des alcanes de même
masse molaire, mais nettement inférieure à celles des acides carboxyliques. A la différence de ces derniers, il n'y a pas d'association par liaison hydrogène.
On le prépare par réaction entre le dichlore et le monoxyde de carbone.
Le phosgène est utilisé comme produit de base dans l'industrie chimique en raison de sa grande réactivité. Un exemple d'application est la synthèse de la cétone de Michler.
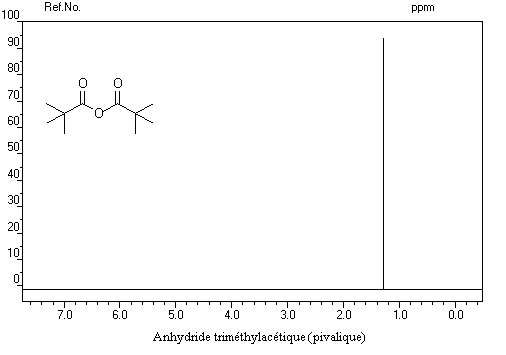
Spectroscopie de RMN Dérivés importants Anhydride acétique Anhydride phtalique Anhydride maléique L'anhydride maléique est préparé par oxydation du benzène. C'est un excellent diénophile dans les réactions de
Diels-Alder. Cétènes Le plus simple des cétènes est préparé par déshydratation de l'acide éthanoïque.
L'anhydride acétique peut être préparé par acylation des ions éthanoate par le chlorure d'éthanoyle.
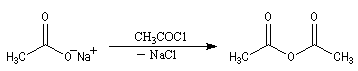
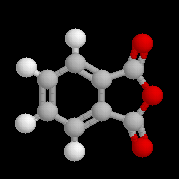
L'anhydride phtalique est préparé par déshydratation de l'acide phtalique à 200 °C.
La réaction avec le glycérol permet la préparation de résines glycérophtaliques. Ces polymères tridimensionnels, entrent dans la composition des peintures et des vernis.
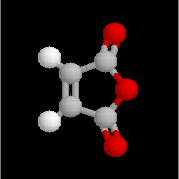
Ce composé esr utilisé pour réticuler les résines polyesters. L'hydrolyse suivie d'hydrogénation fournit l'acide succinique.
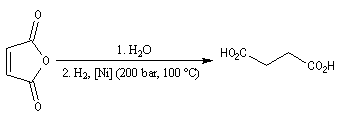
![]()
Etat naturel
Cantharidine
La cantharidine est un composé de type terpénoïde comportant une structure tricyclique.
Ce composé, longtemps réputé pour ses vertus aphrodisiaques a été extrait au début du 19ème siècle d'un scarabée (spanish fly)
par Robiquet.
Malgré les apparences, sa synthèse n'est pas chimiquement évidente car une réaction de Diels-Alder entre le furane et le dérivé diméthylé de l'anhydride maléique ne convient pas car l'équilibre est très défavorable au produit.
Réduction des dérivés d'acides
Réduction en aldéhyde Réactions d'additions-éliminations au niveau du carbonyle
La réaction de Rosenmund était autrefois la seule méthode permettant la réduction des chlorures d'acyles en aldéhydes. Elle consiste à utiliser le dihydrogène en présence de Pd de Lindlar (Pd précipité sur un support de sulfate de baryum et empoisonné par PbOAc2 et la quinoléine). Il est moins actif que Pd/C.
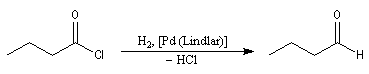
![]()