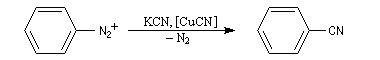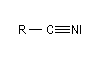
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Les nitriles
Introduction
Généralités
On appelle nitriles les composés comportant le groupe cyano lié à une chaîne alkyle ou phényle. Ils ont la formule générale suivante.
Les nitriles étaient appelés naguère cyanures organiques. On peut considérer qu'ils dérivent formellement du cyanure d'hydrogène par remplacement d'un atome d'hydrogène par un groupe alkyle ou phényle. Cette manière de voir est illustrée par la nomenclature radicofonctionnelle qui rappelle celle des dérivés halogénés. Il faut faire attention que l'analogie avec cette classe de composés s'arrête la. Bien qu'ils donnent lieu à des réactions diverses et variées, les nitriles ne sont pas des composés très réactifs comme en témoigne l'utilisation de certains d'entre-eux comme solvants.
|
|
Le mandélonitrile est un hydroxynitrile dont l'un des énantiomères est dessiné ci-dessous. 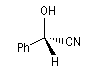 La fonction alcool du mandelonitrile peut former un acétal en se liant à deux molécules de glucose. Le glucoside appelé amygdaline, est présent à l'état naturel dans les amandes amères de plusieurs fruits (pêche, abricot) [25]. |
En milieu basique, le mandélonitrile est instable et sa décomposition fournit l'ion cyanure et du benzaldéhyde. En milieu biologique, la décomposition de l'amygdaline aboutit au même résultat grâce à des réactions catalysées par une enzyme : l'hydroxynitrile lyase.
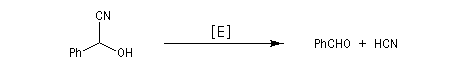
Ces réactions sont mises à profit comme moyen de défense par certains animaux comme les mille-pattes. Le cyanure d'hydrogène libéré est un composé très toxique qui inhibe l'action d'une enzyme intervenant dans la chaîne respiratoire : le cytochrome oxidase a-a3. Le cyanure d'hydrogène a été isolé par le chimiste suédois C. Scheele en 1782 à partir du Bleu de prusse. Il décédera quelques années plus tard, la santé vraisemblablement altérée par le contact répété avec les dérivés du cyanure.
|
|
L'éthanenitrile est appelé acétonitrile dans l'industrie. On peut le préparer par déshydratation de l'éthanamide. On peut aussi réaliser une réaction de substitution nucléophile des ions chlorure du monochlorométhane par les ions cyanure. L'éthanenitrile est utilisé comme solvant industriel sélectif dans l'extraction des cires et des graisses. |
|
|
Le benzonitrile ou cyanure de phényle peut être obtenu à partir du diazonium de l'aniline par réaction de type Sandmeyer.
Il est utilisé comme solvant et comme intermédiaire de synthèse en chimie pharmaceutique. |
|
|
Le propènenitrile ou acrylonitrile a été synthétisé en 1893 par C. Moureu. Il existe plusieurs méthodes de synthèse. L'oxydation du propène en présence d'ammoniac a été mise au point par les chercheurs de la firme Sohio.
Il est utilisé dans la fabrication de fibres textiles (Orlon) et dans la préparation de résines en copolymérisation avec le buta-1,3-diène et le styrène (ABS). |
L'acrylonitrile est le plus simple des nitriles a, b-insaturés. Les composés de cette famille constituent d'excellents diénophiles pour la réaction de Diels-Alder. Le tétracyanoéthène (TCNE) qu'on peut considérer comme un dérivé substitué du précédent, est tellement réactif qu'il donne une réaction de ce type avec l'anthracène.
Nomenclature
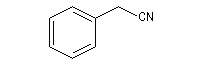
Substitutive Ethanenitrile But-3-ènenitrile Hexanenitrile Hexanedinitrile Cyclohexanecarbonitrile 1,3,6-Hexanetricarbonitrile Radicofonctionnelle Cyanure de phényle Cyanure de benzyle Beaucoup de nitriles sont nommés à partir de noms triviaux formés à partir de celui de l'acide correspondant. Propriétés physiques Puisque l'atome d'azote est plus électronégatif que l'atome de carbone, le groupe cyano est polarisé. On peut rendre compte de cette polarisation par la méthode de la résonance. On remarque l'analogie avec la structure du groupe carbonyle. Constantes physiques Composé M TE sol (g/100 mL H2O) (20 °C, 1 bar) CH 41 82 miscible liq CH 55 97 10 liq CH 69 117 3 liq Du fait d'un moment dipolaire élevé, les nitriles ont des températures d'ébullition légèrement plus grandes que celle des alcools de masse molaire comparable. La miscibilité avec l'eau de l'éthanenitrile s'interprète par la formation de liaisons hydrogène. Spectroscopie Spectroscopie infrarouge La vibration d'élongation de la triple liaison carbone-azote apparaît dans la même zone que celle de la triple liaison carbone-carbone des alcynes. Groupe alcyne nitrile s (cm-1) 2100 - 2140 2200 - 2300 L'intensité est beaucoup plus grande que pour une triple liaison carbone-carbone du fait d'un moment dipolaire important. Spectroscopie RMN En RMN du 13C, le carbone du groupe fonctionnel sort entre 110 ppm et 125 ppm. Il est donc plus déblindé que l'atome de carbone d'un alcyne qui sort entre 65 et 85 ppm. Cela vient du fait que l'atome d'azote est plus électronégatif que l'atome de carbone et il draine une partie de la densité électronique. Réactions d'hydratation Hydratation en amide La réaction est catalysée en milieu acide. Avec les nitriles aromatiques, l'hydrolyse peut être arrêtée au stade de l'amide en opérant en présence d'acide sulfurique concentré à une température suffisamment basse. Une autre méthode d'hydratation sélective des nitriles en amides consiste à utiliser une solution aqueuse d'eau oxygénée en milieu basique [1].
Hydrolyse des nitriles De même, l'hydrolyse des aminonitriles est utilisée dans la synthèse de Strecker des aminoacides. Alcoolyse Selon les conditions expérimentales, les imidates conduisent à plusieurs types de composés :
Réactions de réduction Réduction en aldéhyde par Al(i-Bu)2H
C'est la nomenclature recommandée par l'UICPA. Le composé est nommé en ajoutant nitrile au nom de l'hydrocarbure possédant le même nombre d'atomes de carbone. L'atome de carbone lié à l'azote porte le numéro 1.
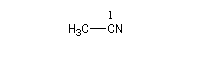
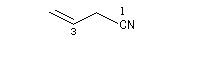
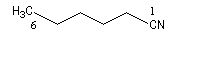

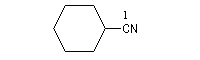
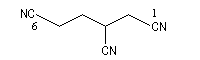
Les composés sont nommés comme des cyanures d'alkyle.
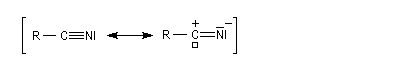
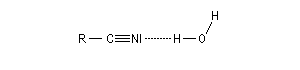
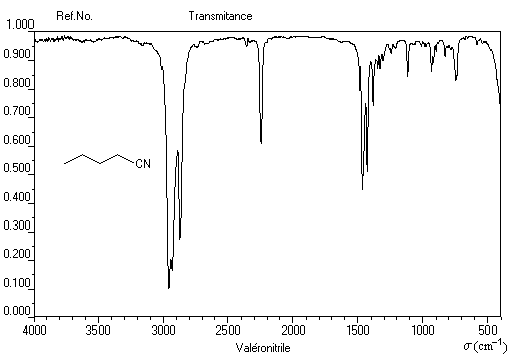
Le spectre infrarouge ci-dessous est celui du pentanenitrile (valéronitrile).
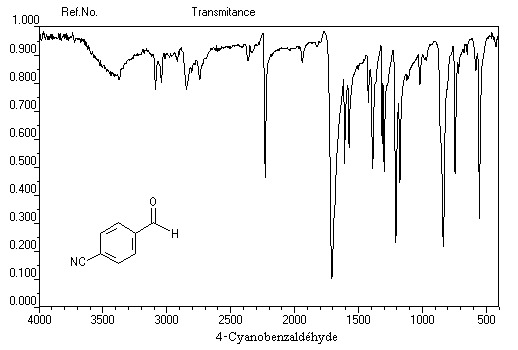
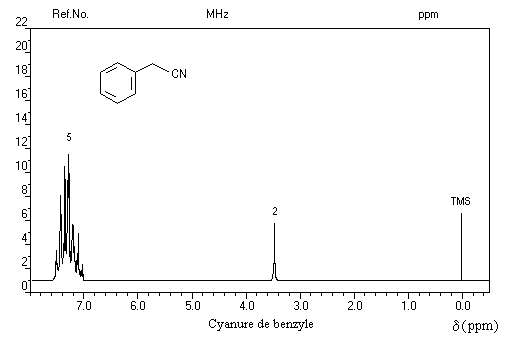
Les protons voisins d'un groupe cyano sont pratiquement déblindés de la même façon que ceux voisins d'un groupe carboxy d'acide carboxylique aux alentours de vers d = 3,5 ppm.
La déshydratation d'un amide sous l'action d'agents déshydratants comme PCl5, P4O10, SOCl2. conduit à un nitrile. Inversement l'hydratation d'un nitrile
fournit l'amide.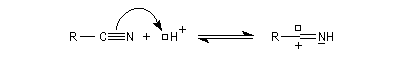
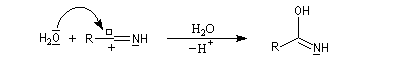
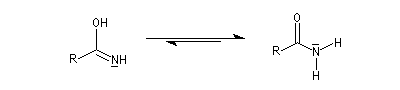
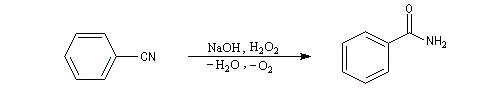
L'hydratation d'un nitrile fournit dans un premier temps l'amide. Si l'on ne prend pas de précautions particulières, la transformation se poursuit par l'hydrolyse de l'amide. Selon la nature du milieu, on obtient l'acide carboxylique ou bien l'ion carboxylate.
L'hydrolyse d'un hydroxy-nitrile comme le mandélonitrile fournit un hydroxy-acide, l'acide mandélique.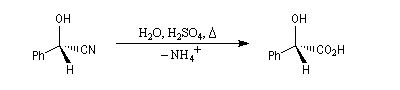
L'alcoolyse s'effectue en traitant le nitrile par un alcool en présence de chlorure d'hydrogène. On obtient un imidate (iminoéther).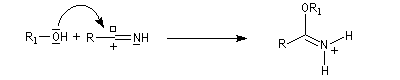
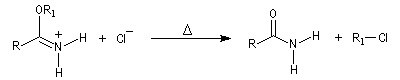
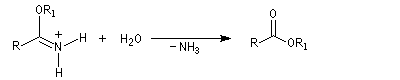
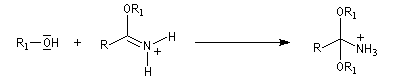
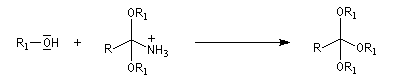
Les nitriles sont réduits en aldéhydes par le bis (2-méthylpropyl) aluminium Al(i-Bu)2H (diisobutylaluminium : DIBAL-H), un hydrure neutre, à - 78 °C. Une température basse est importante afin de prévenir une réduction plus poussée. Le réactif est un réducteur neutre, électrophile du fait de son caractère d'acide de Lewis.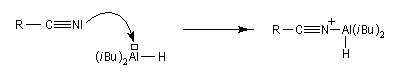
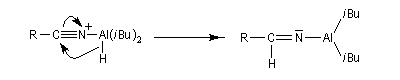
En présence d'eau, on obtient une imine .
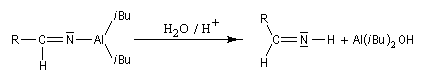
L'hydrolyse de cette imine fournit l'aldéhyde.
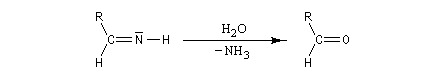
Réduction en amine par H2
La réduction par le dihydrogène est réalisable par catalyse hétérogène. Les catalyseurs utilisés sont les mêmes que pour l'hydrogénation des composés éthyléniques : Rh, Pd, Pt/C ou PtO2. La température est comprise entre 5 °C et 150 °C et la pression entre 1 et 100 bar. La réduction conduit aux amines primaires, secondaires ou tertiaires via une imine intermédiaire. La formation des amines primaires est favorisée par l'utilisation d'un solvant comme l'anhydride acétique en présence d'une forte pression en H2.
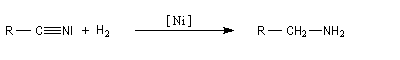
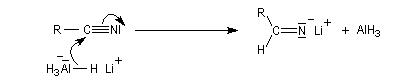
Réduction en amine par LiAlH4
LiAlH4 réduit les nitriles en iminoalanate. Ici plusieurs ions hydrure sont transférables et la réduction se poursuit jusqu'au stade de l'aminoalanate.
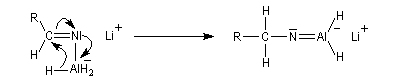
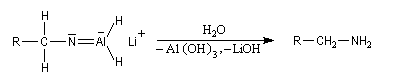
Réactions avec les organométalliques
Organomagnésiens
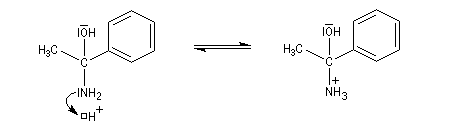
L'addition d'un organomagnésien sur un nitrile conduit à la formation d'un sel d'immonium. Ce sel d'imine bien qu'insaturé ne réagit pas avec le magnésien car cela impliquerait deux charges négatives sur le même atome d'azote.
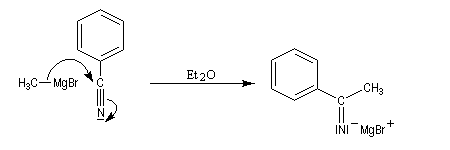
En milieu acide, on obtient une imine non substituée à l'azote.
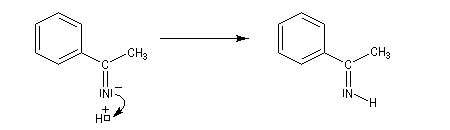
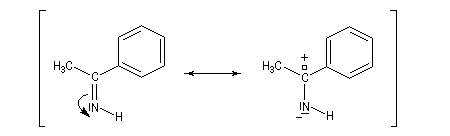
Les imines non substituées sont facilement hydratées en aminoalcools.
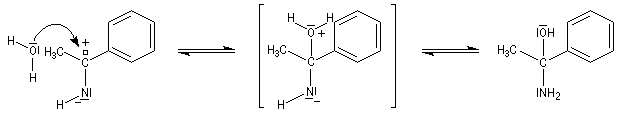
L'aminoalcool obtenu est instable et il est facilement hydrolysé.
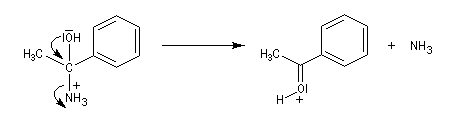
Le produit final est une cétone.
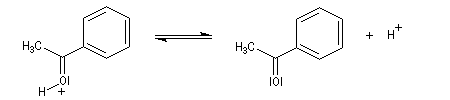
Cette réaction qui constitue une méthode de synthèse des cétones, possède une limitation dans le cas où l'atome d'hydrogène situé sur l'atome de carbone en a de la fonction nitrile est trop acide. Il est alors arraché par l'organométallique. De nos jours on utilise plus volontiers la réaction entre un organométallique et un amide de Weinreb.
Organolithiens
L'exemple suivant concerne la réaction entre le phénylithium et l'éthanenitrile.
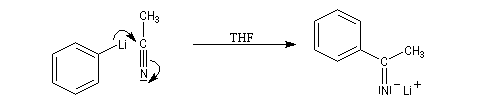
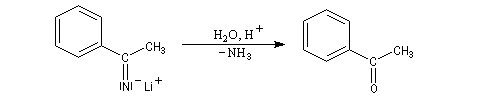
Propriétés des atomes d'hydrogène en a du groupe cyano
Mobilité protonique
Comme chez les composés carbonylés, la mobilité protonique en a du groupe cyano se manifeste par plusieurs phénomènes :

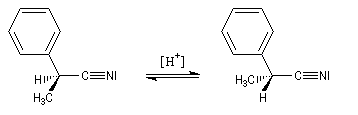
Ces réaction s'interprètent par l'existence d'un équilibre de tautomérie comparable à la tautomérie céto-énolique catalysé par les acides et par les bases.
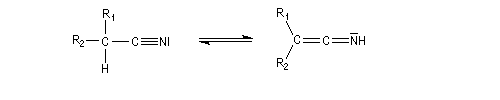
L'atome central est digonal comme dans un cétène.
Préparation de carbanions
La préparation de carbanions à partir des nitriles s'effectue de la même façon que la préparation des ions énolate de composés carbonylés. On utilise une base très forte comme le LDA. Puisqu'il n'y a qu'un seul atome de carbone en a, le problème de la régiosélectivité ne se pose pas.
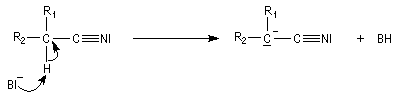
L'anion obtenu ressemble à un ion énolate.
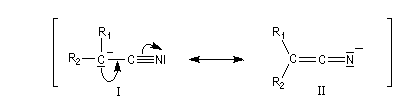
Des carbanions stabilisés par une fonction nitrile sont utilisés dans des variantes de la synthèse malonique.
Alkylation
Elle est comparable à l'alkylation sur l'atome de carbone en a des composés carbonylés.
L'éthanenitrile peut être déprotoné de façon quantitative par le LDA.
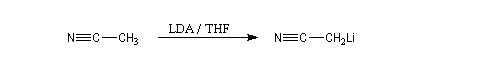
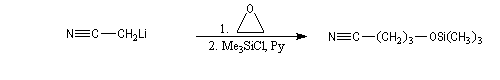
On peut aussi utiliser comme base l'amidure de sodium dans l'ammoniac liquide.
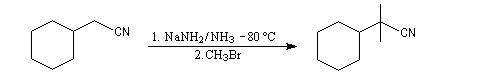
Lorsque l'anion obtenu peut être stabilisé par résonance, on peut utiliser une base moins forte. La réaction peut être réalisée dans des conditions douces en utilisant une catalyse par transfert de phase. Le mode opératoire de la synthèse suivante est donnée par le chimiste polonais M. Makosza l'un des inventeurs de ce type de catalyse [29].
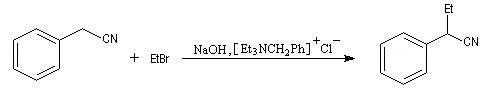
Addition nucléophile
L'addition nucléophile d'un carbanion stabilisé par une fonction nitrile sur un groupe carbonyle, ressemble à la réaction d'aldolisation. On obtient un b-hydroxynitrile qui après déshydratation fournit un nitrile a, b-insaturé.
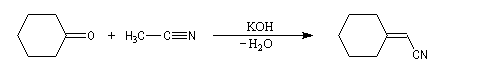
Les nitriles a, b-insaturés, sont des substrats utiles pour les réactions de Michaël. Un exemple de réalisation expérimentale est donné à la référence [24].
Cyclisation de Thorpe
La cyclisation de Thorpe est une réaction de condensation des dinitriles. Elle consiste en l'addition nucléophile d'un carbanion formé en a d'une première fonction nitrile sur une seconde.
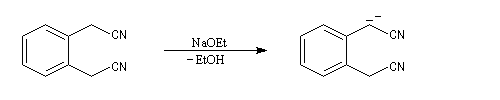
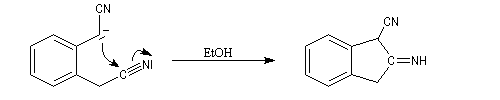
L'imine non substituée formée est hydrolysée et l'on obtient finalement un composé carbonylé.
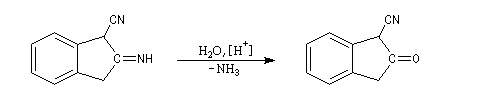
Cette réaction est à rapprocher de la cyclisation de Dieckmann.
Une variante de la réaction de Thorpe, appelée réaction de Ziegler, consiste à effectuer la cyclisation de dinitriles à haute dilution dans l'éthoxyéthane en présence d'un amidure alcalin.
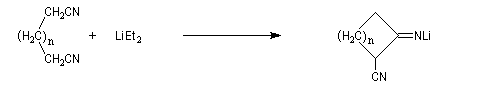
L'imino-nitrile obtenu est ensuite hydrolysé.
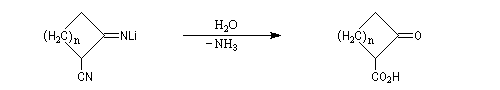
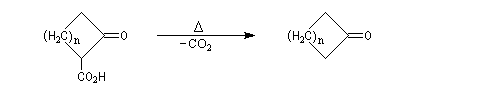
Propriétés nucléophiles de l'azote
Réaction de Ritter
La réaction de Ritter permet de passer des nitriles aux amides N-substitués.
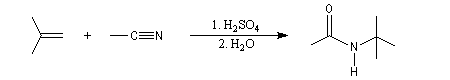
Elle débute par l'addition nucléophile de l'azote du nitrile sur un carbocation généré dans le milieu réactionnel à partir d'un alcool tertiaire ou d'un composé éthylénique.
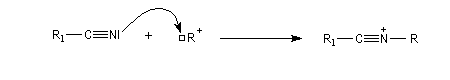
L'ion formé est stabilisé par résonance.
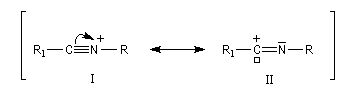
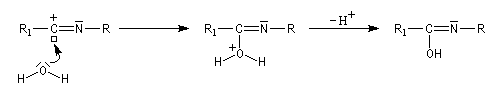
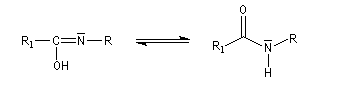
L'hydrolyse de l'amide permet d'obtenir une amine. Lorsqu'elle est suivie d'une hydrolyse de l'amide formée, la réaction de Ritter constitue une méthode de préparation des alkylamines tertiaires. Celles-ci ne peuvent être préparées valablement par alkylation d'Hofmann de l'ammoniac car on obtient un trop grand pourcentage d'élimination.
Ouvrages expérimentauxOuvrages théoriques
A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
J. March - Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
V. Minkine - Théorie de la structure moléculaire, Editions Mir 1979.
N. T. Anh - Introduction à la chimie moléculaire, Ellipses 1994.
R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
P. Atkins - Molecular Quantum Mechanics, Oxford University Press 1983.
J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.
P. Chaquin, F. Volatron, Chimie organique : une approche orbitalaire de Boeck 2015
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
novembre 2016